Article Text

Abstract
Introduction: Clinical variability associated with the common 22q11.2 microdeletion is well known, and has led to a broad application of FISH diagnostics with probes for loci TUPLE1 or D22S75 (N25), although, rarely reported atypical deletions associated with the same phenotypic spectrum would not be discovered by these probes. As most types of 22q11.2 deletions occur between low copy repeats within the region (LCR22), we assumed that atypical deletions should be more common than has been reported. To address this question and the possibility of a deletion size related genotype-phenotype correlation, we systematically assessed the frequency of typical and atypical 22q11.2 deletions in a large cohort of patients.
Methods: We used a set of 10 fluorescent in situ hybridisation (FISH) DNA probes, capable of detecting all reported and hypothetical deletions between the LCR22, and analysed 350 patients. Deletion sizes in atypical deletions were established by use of further FISH probes. Frequency of certain atypical deletions was analysed in controls by FISH and quantitative PCR.
Results: Patients with conotruncal heart defects (ctCHD) and with typical VCFS phenotype showed the common 3 Mb or nested 1.5 Mb deletions (in 18.5% and 78.6%, respectively), but no atypical deletion, while 5% (3/63) of patients with a mildly suggestive, atypical phenotype showed atypical distal deletions, which were not detected in patients with mental retardation of unknown origin or in healthy controls.
Discussion: These statistically significant differences demonstrate that atypical distal 22q11.2 deletions are very uncommon in patients with ctCHDs, while atypical congenital heart defects and mild dysmorphism are recognisable feature of atypical distal deletions. Further phenotype-genotype analysis disclosed association of significant developmental delay with the distal part of the common deletion region, and choanal atresia and atypical CHDs with the adjacent distal deletion region.
- ctCHD, conotruncal congenital heart defects
- DGS, DiGeorge syndrome
- FISH, fluorescent in situ hybridisation
- LCR, low copy repeat
- MR/MCA, mental retardation/multiple congenital anomalies
- PDA, persistent ductus arteriosis Botalli
- PFO, persistent foramen ovale
- SNP, single nucleotide polymorphism
- VCFS, velocardiofacial syndrome
- VSD, ventricular septal defect
- 22q11.2 atypical deletions
- affected sib pair
- congenital heart defects
- mental retardation
- choanal atresia
Statistics from Altmetric.com
- ctCHD, conotruncal congenital heart defects
- DGS, DiGeorge syndrome
- FISH, fluorescent in situ hybridisation
- LCR, low copy repeat
- MR/MCA, mental retardation/multiple congenital anomalies
- PDA, persistent ductus arteriosis Botalli
- PFO, persistent foramen ovale
- SNP, single nucleotide polymorphism
- VCFS, velocardiofacial syndrome
- VSD, ventricular septal defect
- 22q11.2 atypical deletions
- affected sib pair
- congenital heart defects
- mental retardation
- choanal atresia
The well known recurrent ∼3 Mb microdeletion within chromosomal band 22q11.2 is associated with a variety of symptoms and syndromes,1–3 including DiGeorge and velocardiofacial syndromes (DGS/VCFS; OMIM 188400), and constitutes a major cause of congenital heart disease, accounting for about 5% of all such defects in live born children.4 Despite variable phenotypes even within families,3,5 84–90% of patients with VCFS and deletions of N25/TUPLE1 show an identical deletion size of approximately 3 Mb, while 7–14% have a proximally nested ∼1.5 Mb deletion.6–8 Furthermore, Kurahashi et al9 investigated 100 patients with several fluorescent in situ hybridisation (FISH) probes from the 3 Mb region and detected a small distally nested deletion in one patient. A few patients with typical phenotype but normal results for the commonly used probes were further investigated and reported to have atypical nested deletions,10–14 but also adjacent atypical distal deletions.15,16 As both common and atypical deletions were shown to be mediated by several low copy repeats (LCR) within the region,6,15,17,18 we suspected that, if investigated systematically, atypical deletions would be revealed to be more common than reported. Thus, using a set of 10 FISH probes that would detect all reported and hypothetical types of deletions between the LCRs, we analysed 350 patients with symptoms of the DGS/VCFS spectrum.
SUBJECTS AND METHODS
The study was approved by the university ethics review board and appropriate informed consent was obtained from all participants or their parents.
Patients
We initially analysed 73 patients with conotruncal congenital heart defects (ctCHD) such as interrupted aortic arch, truncus arteriosus communis, Fallot tetralogy, or pulmonary atresia, recruited from the paediatric cardiology centres of the universities of Erlangen and Tuebingen. The patients had previously showed normal results for the FISH probe N25 (D22S75).
We later prospectively analysed 200 patients with ctCHD. Clinical details of these patients have been published elsewhere or are in preparation.19–23 While the latter patients were recruited only for the characteristic heart phenotype, 77 further patients without characteristic ctCHDs were analysed because clinical investigations at the genetics clinic requested due to developmental delay and/or malformations (mental retardation/multiple congenital anomalies; MR/MCA) of unknown origin revealed features consistent with the 22q11.2 deletion spectrum. Inclusion criteria for the latter were either developmental delay, short stature, or frequent infections in addition to at least two minor facial anomalies compatible with the DGS/VCFS spectrum, or the presence of at least one minor dysmorphism in addition to one of the following: CHD, urogenital anomaly, cleft palate, choanal atresia, hypocalcaemia. Only 14 of these latter patients were clinically judged as having a typical DGS/VCFS facial phenotype. In addition, one affected sibling pair from healthy parents known from the German parents’ support group was analysed for deletion size and parental origin.
The control groups comprised up to 300 patients with MR/MCA of unknown origin not meeting the inclusion criteria for the 22q11.2 deletion study, and 285 health controls were analysed for certain deletions.
Methods
FISH analysis was performed on metaphase spreads from cultivated lymphocytes with the following 10 DNA probes covering the common and atypical deletion intervals: 6E8 (D22S427), 109G12 (109G12-1),17 51H3 (D22S1649), Pac 140D4 (TUPLE1/HIRA), D0832 (COMT),8 48c12 (D22S264),18 co23 (UFD1L),24 cHKAD26 (D22S935),25 and BAC 438P22 (D22S425).15 Probes were directly labelled with Cy3 by nick translation with a commercial kit (Roche Corp.) and hybridised with a 22q subtelomeric control probe (GS-98-C4, locus 22QTEL31) directly labelled with FluoroX as described previously.26 In patients with atypical deletions and their parents, appropriate microsatellite markers and either probes RP11-140M6 and RP11-134C5 or a commercial Bcr/Abl probe (ONCOR), RP11-1146O18, and RP11-20P18 were analysed as described previoulsy.27 MR/MCA controls were analysed by FISH with the probes Pac 140D4, cHKAD26 and BAC 438P22. Healthy controls were analysed by quantitative PCR using Taqman assays with minor grove binding probes derived from exon 2 of CRKL and of exon 1 of GNAZ. Primer and probe sequences used were: CRKLe2F2 forward primer(GTGGAGTGCCCGGAACAA), CRKLe2R2 reverse (CTTTTCGACATAAGGGACAGGAAT), CRKLe2_mgb_probe2 (ATGGCCGGGTTGGG), GNAZe1F forward (CGGGCATTGTGGAGAACAA), GNAZe1R reverse (CCCACGTCCACCATCTTGA), and GNAZe1_mgb_probe (CTTCAAGGAGCTCACC). The assays were performed in triplicate, each multiplexed with an albumin probe as an internal reference. Calculation of copy numbers by the ΔΔCt method was performed as described elsewhere.28 For each assay, two patients with deletions in the region under analysis were used as positive controls.
RESULTS
In the 73 retrospective patients with ctCHD with normal results for locus D22S75 no further deletion was detected. A deletion within 22q11.2 was found in 37 of the 200 (18.5%) prospective patients with ctCHD, 11 of the 14 (78.6%) typical VCFS patients and 3 of the 63 (4.8%) patients with symptoms weakly suggestive of VCFS. Of 51 deletions, 43 (84.3%) had the common ∼3 Mb size encompassing probes c51H3, c70A2, P140D4, co23, D0832, c48c12, and cHKAD26, and three (5.9%) had the proximally nested ∼1.5 Mb deletion that included probes c51H3, c70A2, P140D4, co23, and D0832 only (table 1). One proximal deletion in addition to the former probes was proximally expanded into the cats’ eye syndrome region (43101, fig 1). Four deletions (7.8 %) were distal deletions; one included c48c12 and cHKAD26 (patient 77104) thus nesting within the common 3 Mb region and overlapping with one reported deletion,11 and two were distally adjacent to the 3 Mb region, including B438P22 and BCR only in one patient (07604) and additionally 109G12 in another patient (31502), thus overlapping the deletion reported earlier by our group15 (fig 1).
Overview of results in different patient groups
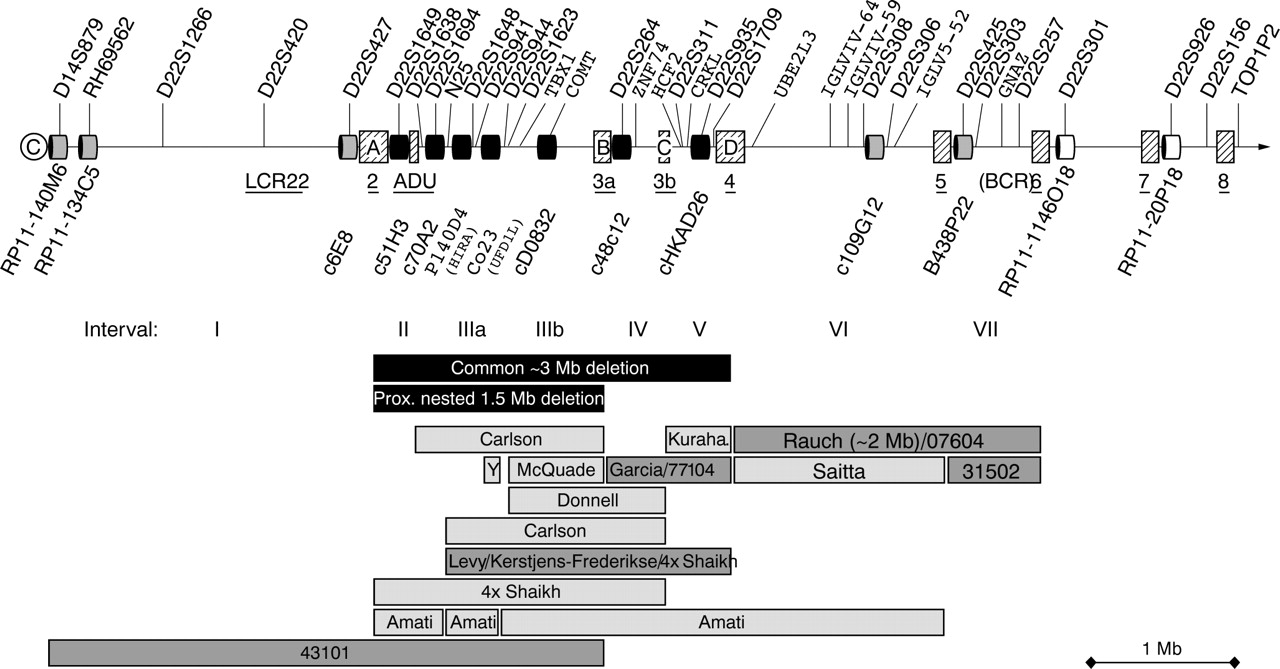
Schematic overview over the DGS/VCFS 22q11.2 deletion region. Low copy repeats are depicted as shaded squares and labelled with LCR22-1 to 8, and ADU according to Edelmann et al18 and Mc Dermid & Morrow,38 and A–D according to Shaikh et al..6 FISH clones used for this study are depicted as black, grey, and white boxes. Bars below the map depict deletion sizes found in this study and in reported cases (light grey).6,7,8,9,10,11,12,13,15,16,39,40,41 Note that exact deletion borders are sometimes only estimated in reported cases.
The common ∼3 Mb deletion were only detected in patients with ctCHD and in patients clinically judged as typical for DGS/VCFS. The proximally nested and enlarged ∼1.5 Mb deletions were seen in four patients with typical ctCHD and one patient with VCFS but with normal development, while all three distal deletions were found in patients with symptoms only weakly suggestive of 22q11.2 deletion. The different frequencies of atypical deletions were statistically significant (table 2). Patient 77104, with the ∼1 Mb nested distal deletion, had been referred for speech delay, but otherwise showed only mild hypotonia, frequent infections, and attacks of pseudocroup, as well as a small mouth, mild retrognathia, and mild ptosis suggestive of DGS/VCFS. At the age of 6 years he was mildly mentally retarded, and suffered from increased anxiety and gave the impression of psychological instability, but formal testing was pending (fig 2C, D; table 3). The deletion was excluded in his mother, but his father, who was said to suffer from schizophrenia, was not available for analysis. Patient 31502, with the ∼0.6 Mb deletion in the BCR region, was referred at the age of 8.5 months because of her valvular pulmonic stenosis with muscular ventricular septal defect (VSD), small PFO, persistent foramen ovale; and PDA, persistent ductus arteriosis Botalli. Her appearance and psychomotor development was normal, and only very careful investigation disclosed minimal broad folding of the right helix and somewhat widely spaced inverted nipples (figure 2G–I). Her healthy father, who had narrow palpebral fissures and low set ears, was shown to carry the same deletion (fig 2J, K). Further relatives of the father were not available for investigation. Patient 07604, with the second atypical distal deletion of about 2.1 Mb in the IGLV region, was referred at the age of 7.5 years with a diagnosis of CHARGE (coloboma, heart anomaly, atresia, retardation, genital anomaly, ear anomaly) syndrome based on bilateral choanal atresia, right sided preauricular tag, hypoplastic irides, small VSD, and conductive hearing loss (fig 2E, F). In addition, she showed frequent infections, strabism, hyperopia, and learning difficulties with a low normal average IQ of 85 (Kaufmann Assessment Battery for children). The deletion was excluded in both healthy parents.
Statistical evaluation of significance of different frequencies of atypical distal deletions by Fischer exact testing
Summary of phenotypic findings according to different atypical small 22q11.2 deletions

{kind=link}
{kind=link}
Facial appearance of patients with typical and atypical 22q11.2 deletions. Written consent for publication of these images was given by individuals or their legal guardians. (A, B) Mild expression of the typical VCFS facial appearance in the sibling pair with the common 3 Mb deletion (A) and the proximally nested 1.5 Mb deletion (B). (C, D) Patient 77104 at 6 years of age. Note the small mouth, pointed nasal tip, mild ptosis and low set ears mildly suggestive of 22q11.2 deletion. (E, F) Patient 07604 at age 7.5 years, showing narrow eyes, thin upper lip, mild hypertelorism and broad, high nasal bridge. (G–I) Patient 31502 at age 8.5 months, showing slightly broad folding of helix, thin upper lip, and widely spaced, inverted nipples. (J, K) Father of patient 31502 with the same deletion showing narrow eyes, somewhat flat nasal tip, thin upper lip, and low set ears.
The atypical distal deletions were not detected in up to 300 patients with MR/MCA of unknown origin not fulfilling the inclusion criteria for the 22q11.2 study nor in 285 healthy controls.
Sizing of the 22q11.2 deletion in the affected sibling pair, neither of whose parents had a deletion, unexpectedly showed a common 3 Mb deletion in the boy and a proximally nested 1.5 Mb deletion in the girl. From the eight polymorphic markers of the deleted regions tested, only D22S941 was fully informative, indicating a paternally derived deletion in the boy and a maternally derived deletion in the girl. D22S1623 was informative for the boy and confirmed a paternally derived deletion. To confirm the different parental origin of the deletions, 23 single nucleotide polymorphisms (SNPs) from the TBX1 genomic region, where both siblings are hemizygous, were analysed and showed divergent alleles at 11 SNPs. While the boy had mild mental retardation, hypocalcemia, scoliosis and frequent infections, the girl attended a mainstream school and had only minor hyperactivity problems, frequent infections, and febrile seizures (figure 2A, B).
DISCUSSION
To date, even with reference to non-overlapping deletions, no consistent genotype phenotype correlation has been shown for 22q11.2 deletions.7,8,9,10,11,12,13,14,16 Nevertheless, our systematic assessment of typical and atypical 22q11.2 deletions in a large number of patients clearly demonstrates a significant correlation between deletion site and phenotypic expression.
Although single case exceptions have been reported, our results reveal that if ctCHD or Shprintzen/VCFS syndromes are related to 22q11.2 deletion, they are usually associated with the common 3 Mb or the frequent 1.5 Mb proximally nested deletions, both encompassing the T box transcription factor TBX1. This is in line with the finding of Yagi et al,29 who revealed a deletion with the proximal N25 probe in 96% of 235 typical DGS/conotruncal anomaly face syndrome patients, but no atypical deletion in the remaining 10 patients investigated with further probes. Moreover, in 3 of these remaining 10 patients, one frameshift and two missense mutations within TBX1 were detected, assigning the main DGS/VCFS phenotype (facial anomaly, conotruncal heart defects, hypoplastic thymus, parathyroid dysfunction, and velopharyngeal insufficiency) to TBX1 haploinsufficiency. Even though the role of TBX1 in heart development was also demonstrated in mouse models,30,31 there is some evidence for additional heart genes within the region such as Crkl,32Hira,33 and Ufd1l.14 Therefore, it seems likely that additional genes contribute to congenital heart defects in patients with atypical deletions excluding TBX1, although positional effects might be possible.
Like the patients with TBX1 mutations, none of our four patients with the proximal 1.5 Mb nested deletion showed severe learning disability or mental retardation, which is found in 38% of 22q11.2 deletion patients.1 Bartsch et al34 reported four patients with assumed similar ∼1.5 Mb deletions who displayed typical DGS/VCFS physical problems, but with both older patients showing normal development. As individual mental development is subject to multifactorial influences including familial background, our finding of correlation of mental development with different deletion size within the same family provides further evidence for this suggestive correlation, as does our patient 77104 with the distally nested deletion, who showed significant developmental delay and possibly early signs of psychiatric illness.35 Learning disabilities were also reported by Garcia et al11 in their case with similar distal deletion.
The unexpected finding of different deletion sizes and different parent of origin within our siblings from parents without deletions further emphasises the necessity of further investigations in affected siblings before counselling for germline mosaicism. Independent de novo 22q11.2 deletions have previously been reported in first cousins.36
The frequency of atypical distal deletions that do not overlap with the common deletion region in our patients with only mildly suggestive features of the DGS/VCFS spectrum differs significantly from the lack of these deletions in 285 healthy controls, in up to 300 patients with unclassified mental retardation, and in 273 patients with ctCHD. Development was normal in the patient and father with the smaller deletion distally nested within the adjacent region (31502). Development was only mildly delayed in our patient 07604 and previously published15 patients with the larger adjacent deletions, and in the previously published patient with the smaller deletion proximally nested within the adjacent deletion region.16 Although exceptions are reported, the type of congenital heart defect in our new patients with deletions distal to the common 3 Mb region is not characteristic for 22q11.2 deletion in the majority of cases,1 and their very mild minor anomalies would usually not be considered relevant. Interestingly, two of the five patients known to have a deletion of the D22S308 region had choanal atresia, compared with only 1% of patients with common deletions.1 As choanal atresia is a major feature of CHARGE syndrome, some of the CHD7 mutation negative patients37 might therefore have an atypical distal 22q11.2 deletion.
It is obvious that phenotypic expression in 22q11.2 deletions is modified by factors other than deletion size, which hampers the identification of their contribution. However, our systematic approach reveals an incompletely penetrant genotype phenotype correlation, with a critical role in ctCHD and the typical DGS/VCFS facial features of the proximally nested interval that contains TBX1, and of the distally nested interval that contains CRKL for major mental impairment. The distally adjacent interval containing D22S308 and/or BCR is frequently associated with choanal atresia and usually atypical congenital heart defects such as arterial stenoses, and the proximal area is associated with mild learning difficulties.
Although the incidence of atypical deletions was considered low and only probes covering the D22S75 or TUPLE1/HIRA loci are generally used for diagnostics, our results show that, by using the latter probes only, about 6% of 22q11.2 deletions are missed. However, efficiency of labour is a consideration, and atypical deletions not detectable with the widely used commercial probes are rare in patients with conotruncal heart defects, but common in patients with atypical phenotypes only weakly overlapping with the DGS/VCFS spectrum.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft grants RA 833/4-1 and 5-1 to AR and HO 1145/2-1 to MH. We thank B Morrow, P Scambler, G Novelli, J Flint, and the Japanese Cancer Resource Bank for providing FISH clones. B Morrow kindly shared LCL22 position data before publication. We are grateful to M Kirsch, L Klassen, and B Dintenfelder for excellent technical assistance. We also thank the families for their kind cooperation.
REFERENCES
Footnotes
-
Competing interests: none declared
-
The study was approved by the University of Erlangen-Nuremberg ethical review board and appropriate informed consent was obtained from human subjects. In individuals shown by photographs written consent for publication of images were given by individuals or their legal guardians.