Article Text

Abstract
Background: Allele variants of COL11A2, encoding collagen type XI α2, cause autosomal dominant non-syndromic hearing loss (ARNSHL) at the DFNA13 locus (MIM 601868) and various syndromes that include a deafness phenotype.
Objective: To describe a genome-wide scan carried out on a consanguineous Iranian family segregating ARNSHL.
Results: Genotyping data identified a novel locus for ARNSHL on chromosome 6p21.3, which was designated DFNB53. Homozygosity for the P621T mutation of COL11A2 was present in all deaf persons in this family; this same variation was absent in 269 Iranian controls. Sequence comparison of collagen type XI α1 and α2 peptides across species shows that the replaced proline is an evolutionarily conserved amino acid.
Conclusions: The P621T mutation of COL11A2 affects the Y position of the canonical -Gly-X-Y- repeat in collagens. It lies near the amino-terminus of the triple helical region and causes ARNSHL. This finding suggests that mutation type and location are critical determinants in defining the phenotype of COL11A2 associated diseases.
- ARNSHL, autosomal recessive non-syndromic hearing loss
- OSMED, otospondylomegaepiphyseal dysplasia
- PCR–RFLP, polymerase chain reaction–restriction fragment length polymorphism
- collagen
- non-syndromic hearing loss
- DFNB53
- COL11A2
Statistics from Altmetric.com
- ARNSHL, autosomal recessive non-syndromic hearing loss
- OSMED, otospondylomegaepiphyseal dysplasia
- PCR–RFLP, polymerase chain reaction–restriction fragment length polymorphism
Hearing loss is the most common sensory impairment. It affects one of every 500 newborn infants,1,2 and another one of every 1000 children becomes severely or profoundly hearing impaired before adulthood.3 In most cases, additional phenotypically distinguishing signs and symptoms are absent, consistent with the diagnosis of non-syndromic hearing loss (NSHL). Over the past decade, researches have mapped 92 loci and cloned 41 genes associated with non-syndromic hearing loss inherited in a Mendelian dominant, recessive, or X linked fashion (hereditary hearing loss homepage, http://www.uia.ac.be/dnalab/hhh/).
As the main components of bone and cartilage, collagens are related intimately to auditory function and, not surprisingly, mutations in collagens are found in many syndromic types of hearing loss. Included in this list are osteogenesis imperfecta (MIM 166200, 166220, 259420, COL1A1, and COL1A2), Alport syndrome (MIM 301050, 203780, 104200, COL4A3, COL4A4, and COL4A5), otospondylomegaepiphyseal dysplasia (OSMED syndrome)(COL11A2), and Stickler syndrome in both its classic (STL1, MIM 108300, COL2A1; STL2, MIM 604841, COL11A1) and non-ocular forms (STL3, MIM 184840, COL11A2), although the latter is more appropriately called heterozygous OSMED syndrome. Mutations in COL11A2 also cause autosomal dominant non-syndromic hearing loss at the DFNA13 locus.4
The collagen family is comprised of 19 collagens encoded by at least 32 unique genes.5 The common signature collagen motif is the sequential repetition of the amino acid triplet -Gly-X-Y-, where many of the X- and Y- positions are filled by the ring amino acids proline and hydroxyproline, facilitating the intertwining of three collagen polypeptide chains into a triple helix.6 Type XI collagen is a minor collagen, accounting for less than 10% of total cartilage collagen. It functions as a spacer, maintaining the interfibrillar distance and fibril diameter of type II collagen.7,8 The three α chain polypeptides of type XI triple helix are α1(XI), α2(XI), and α3(XI) and are encoded by COL11A1 (1p21), COL11A2 (6p21.3), and COL2A1 (12q13.11–q13.2), respectively.9
All diseases associated with allele variants of COL11A2 including non-ocular Stickler syndrome, OSMED syndrome and DFNA13, have a hearing impairment phenotype. The mutations of COL11A2 associated with these diseases include missense mutations, nonsense mutations, small deletions, and insertions.10–15 Phenotype–genotype comparisons suggest that the different phenotypes are mutation dependent. As collagen folding begins at the carboxy terminus where the nucleation domain is located, mutations close to the carboxy terminus generate a more severe phenotype.16 This positional effect of missense mutations is common to other collagen associated diseases. For instance, a glycine to asparagine substitution in exon 22 of COL2A1 causes lethal chondrodysplasia,17 while the same amino acid substitution in exon 10 causes only isolated ocular abnormalities.18 A nonsense or frameshift/stop mutation, in comparison to a missense mutation, is frequently silent in the heterozygote state.
Here we describe the identification of a missense mutation in COL11A2 in a consanguineous Iranian family segregating autosomal dominant non-syndromic hearing loss (ARNSHL). The mutation does not affect a canonical glycine of the -Gly-X-Y- triplet repeat motif and neither is it close to the carboxy terminus. Our finding supports the hypothesis that the clinical phenotypes of COL11A2 associated diseases are a reflection of mutation type and location.
METHODS
Patients and family
L-622 family members were ascertained in a combined otolaryngology/genetics clinic in Iran. A detailed history was taken to exclude conditions such as rubella, prematurity, drug use during pregnancy, perinatal trauma, sudden infant death syndrome, and meningitis. Persons with hearing loss underwent a complete physical examination, with ancillary tests as required. Otoscopy was done to exclude middle ear disease, followed by audiometry to quantitate the degree of hearing loss. On consenting persons, venous blood samples were obtained for DNA extraction. All procedures were approved by the institutional review boards of the University of Social Welfare and Rehabilitation Sciences, Tehran, Iran and the University of Iowa, Iowa City, Iowa USA.
Microsatellite genotyping in the L-622 family
A genome-wide scan was done using 400 fluorescent dye labelled microsatellite markers with an average spacing of 10 cM across the 22 autosomes and chromosome X (Prism Linkage Mapping Set, version 2.5; Applied Biosystems, Foster City, California, USA). Microsatellite markers were amplified by polymerase chain reaction (PCR) and analysed on an ABI Prism 3100 genetic analyser. Alleles were assigned using GeneMapper 3.0 software (Applied Biosystems). Haplotype reconstruction was determined with custom-made GeneScan software.
Mutation analysis by sequencing genomic DNA
All exons of COL11A2 and their flanking sequences were amplified by PCR and sequenced using an ABI model 3700 automated sequencer. Sequence data were compared with published sequence of COL11A2 using the Sequencher 4.1.4 software program package (Gene Codes Inc). Mutation position was assigned according to mRNA sequence from GenBank NM_080680 with nt 1 as first nucleotide of start codon.
PCR–RFLP genotyping of the missense mutation
Primers flanking exon 21 were used to PCR amplify genomic DNA of all participating L-622 family members and 269 Iranian controls. PCR products were digested with BsrI to yield either a 218 base pair (bp) fragment (non-mutant allele) or 116+102 bp fragments (mutant allele), which were resolved by 2% agarose gel electrophoresis.
GenBank accession numbers
Accession numbers included COL11A2 DNA, U32169; mRNA, NM_080680; collagen α1(XI), NP_542196 (Homo sapiens), NP_031755 (Mus musculus); collagen α2(XI), NP_542411 (Homo sapiens), NP_034056 (Mus musculus), NP_997693 (Rattus norvegicus), and AAH43799 (Xenopus laevis).
RESULTS
Clinical phenotype
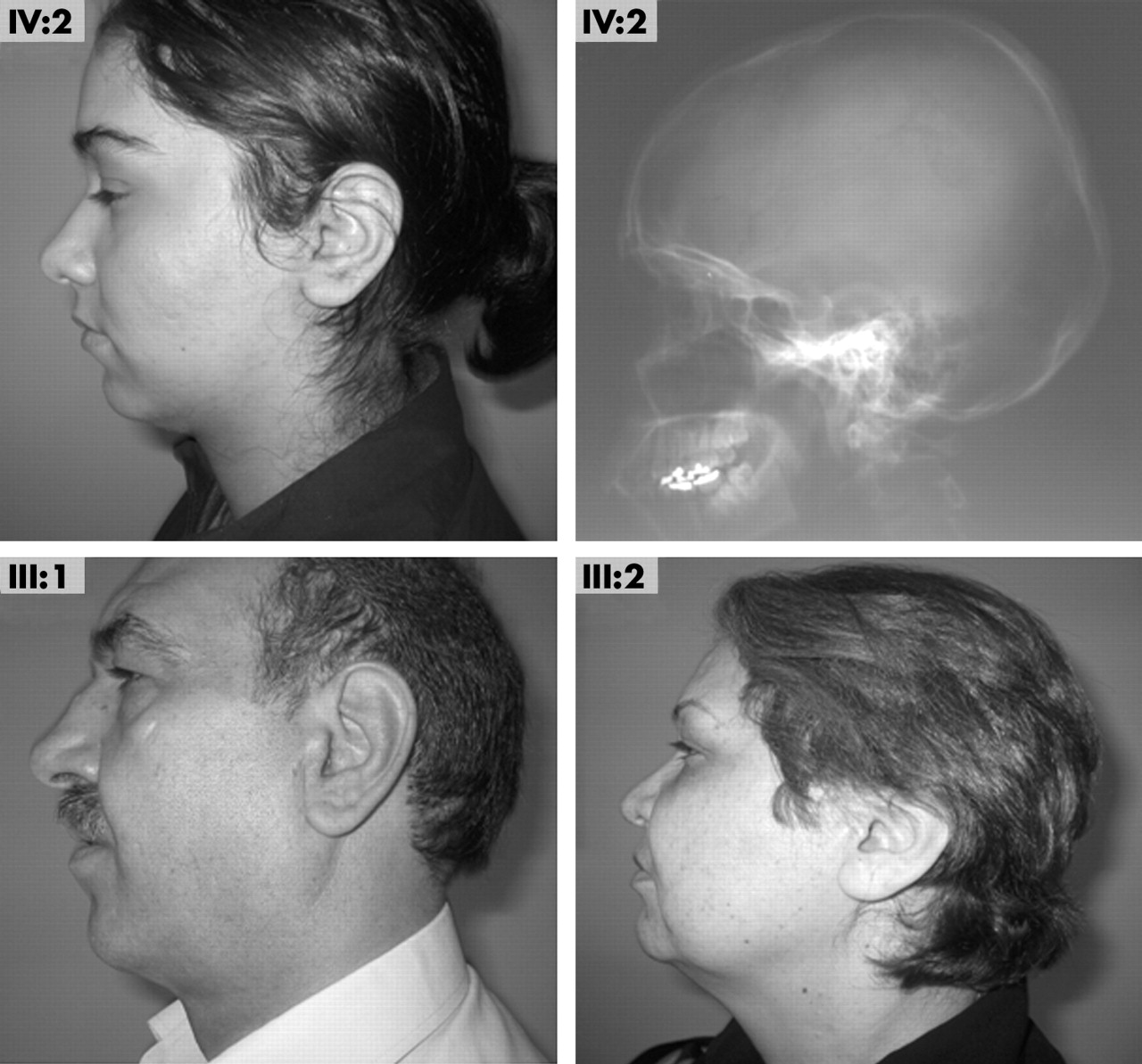
Clinical findings in family L622, a consanguineous Iranian family, were consistent with the diagnosis of ARNSHL (fig 1). All affected individuals had a history of prelingual profound hearing loss, with normal vestibular function as evidenced by age appropriate developmental motor milestones and physical tests of balance (Romberg test and heel–toe walking). Pure tone audiometry confirmed the presence of non-progressive profound sensorineural hearing loss (fig 1). Ophthalmological examination done by an ophthalmologist on all affected family members ruled out evidence of myopia, vitreo-retinal detachment, cataract, glaucoma, or other eye symptoms. An otolaryngological examination carried out by an otolaryngologist excluded midface hypoplasia and obvious palatal clefting, while palpation of the secondary palate excluded the presence of a submucous cleft palate. Stature was normal. In two of the five affected persons (V:2 and V:3) a general bone survey including the thoracolumbar spine, wrists, and knees showed no evidence of joint degeneration, abnormal epiphyseal development, or bone dysplasia. The profile of IV:2 showed mild mandibular retrognathia, consistent with a lateral skull radiograph, which additionally showed normal maxillary development and a class II dentoskeletal relationship. The father of IV:2 also had mild mandibular retrognathia (fig 2). Non-affected individuals had normal hearing as documented by pure-tone audiometry (fig 1).

The L622 pedigree showing haplotypes over the linked region on chromosome 6. The arrow (V:1) indicates an obligate ancestral recombination event between D6S276 and D6S1610 on the maternal chromosome. Individuals shown in black have profound sensorineural hearing loss as indicated by pure tone audiometry. III:2 and IV:5 have normal hearing excluding a mild phenotype in carriers of the P621T allele variant of COL11A2. IV:3, who is homozygous for the wild-type allele, also has normal hearing. Audiometry was not available for other persons in the pedigree (audiograms: ○, right ear; X, left ear).

Lateral skull radiograph on IV:2 shows normal maxillary development, a class II dentoskeletal relationship, and mild mandibular retrognathia. Mandibular retrognathia is seen on a profile view and is present in III:1 but not in III:2 (parents of IV:2), both of whom are carriers of the P621T allele variant of COL11A2. (Photographs reproduced with patients’ consent.)
Localisation of DFNB53 to 6p21.3
We completed a genome-wide linkage analysis using 400 polymorphic microsatellite markers (Applied Biosystems) on 11 members of family L622 (fig 1). Genotyping data from subfamily A identified a novel locus for ARNSHL on chromosome 6p21.3 flanked by D6S422 (telomeric) and D6S257 (centromeric), which was designated DFNB53. Individual V:1 further narrowed the DFNB53 interval to a 9.4 cM region flanked by D6S276 (telomeric) to D6S1610 (centromeric).
Mutation analysis in family L622
The 9.4 cM DFNB53 interval includes 246 known genes, including COL11A2. Because mutations in COL11A2 cause deafness at the DFNA13 locus, we considered it an excellent functional candidate and completed mutation screening of its 66 exons. We identified a cytosine to adenine transition in exon 21 (nt 1861) in the heterozygous state in individual V:1 and in the homozygous state in individual IV:2 (fig 3A). Further analysis by polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) analysis showed all five affected persons were homozygous for this variation, while the four parents and V:1 were mutation carriers (fig 3B). Genotyping data from 269 Iranian controls (538 chromosomes) did not show the same variation, suggesting that it is rare in the general population. The resulting change at the amino acid level—P621T based on UniProt P13942—affects the Y position of the -Gly-X-Y- repeat near the amino-terminus of triple helical region. Sequence comparison of collagen type XI α1 and α2 peptides across species identifies the replaced proline as an evolutionarily conserved amino acid (fig 3C).

Mutation analysis in the DFNB53 family. (A) Sequence data from IV:2 (congenitally deaf), V:1 (carrier of the P621T allele) and a control person. (B) Polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) data following BsrI digestion of amplified DNA. All affected individuals (−/−) have two digestion products of 116 bp and 102 bp; IV:3, not carrying the mutation, has the undigested product of 218bp; five carriers (+/−) have one undigested and two digested products. (C) Alignment of a portion of collagen type XI proteins from various species shows that P621 (arrow) in the collagen triple helical region is conserved across species (conserved amino acids, dark blue background; similar amino acids, light blue background; non-conserved amino acids, white background; boxed sequence, proline-rich region flanking the mutation site).
Analysis of reported COL11A2 mutations
Several mutations in COL11A2 have been reported, including 10 nonsense or frameshift/stop mutations in patients with recessive OSMED syndrome,10,12 three in-frame deletions in dominant non-ocular Stickler syndrome, and four missense mutations that lead to either dominant or recessive disease4,14,15 depending on the type and location of the mutation (fig 4; table 1).
Reported allele variants of COL11A2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The location of reported missense mutations in collagen α2(XI) chain precursor. Graphic view of the domain structure is from the Protein Families Database (UniProt P13942). Note the relation between mutation position/type and phenotype. Carriers of the DFNB53 and OSMED causing mutations have a normal phenotype. Numbers in parentheses indicate amino acid positions in mature collagen α2(XI) protein and were used in previous publications.
DISCUSSION
Type and location of COL11A2 mutations determine disease phenotype
Mutations of COL11A2 cause syndromic and non-syndromic autosomal dominant and recessive hearing loss.10,11,13–15 This phenotypic spectrum is the consequence of both mutation type and location. Syndromic disease is associated with either a dosage or a dominant negative effect. The former is seen with nonsense or frameshift/stop mutations that are associated with nonsense mediated mRNA decay or generate short lived but rapidly degraded truncated proteins.20 In the heterozygous state, these mutations cause haploinsufficiency but no disease phenotype, while in the homozygous state, they result in absence of α2(XI) and autosomal recessive OSMED syndrome. In-frame deletions in COL11A2, in contrast, are translated into shortened mutant proteins that are incorporated, albeit abnormally, into the collagen triple helix. The phenotypic consequence is dominantly inherited non-ocular Stickler syndrome and reflects a dominant negative mechanism of action.
Missense mutations of COL11A2 cause both syndromic and non-syndromic hearing loss. Of the five reported missense mutations, three change the core amino acid glycine of the -Gly-X-Y- consensus repeat (fig 4; table 1). Because G1441E is closest to the carboxy terminus, which is also the site of trimerisation initiation of the collagen helix, this glycine mutation has the most severe phenotypic consequence of the three—it causes autosomal dominant Weissenbacher–Zweymuller syndrome (MIM 277610, non-ocular Stickler syndrome). G808E causes only autosomal dominant hearing loss, and G661R causes autosomal recessive OSMED syndrome.
Hypothesised effect of the P621T mutation
The homozygous mutation we identified, P621T, differs from reported COL11A2 mutations in two respects: it does not involve glycine and it is not close to the carboxy terminus. We hypothesize that this variant is pathological because it either destabilises the triple helix or affects collagen metabolism.
Proline always occupies the X or Y position in collagen and often undergoes post-translational hydroxylation, a modification that stabilises the triple helix. P621T could destabilise the triple helix in two ways. First, isoleucine-620 could repress glycosylation of threonine-621, and when non-glycosylated, threonine cannot stabilise the triple helix.21 In addition, because the region is proline-rich and therefore able to form only weak hydrogen bonds, a proline substitution may destabilise adjacent amino acid interactions22 (fig 3C).
The P621T substitution also could affect collagen α2(XI) metabolism. Enzymatic degradation of type XI collagen may be important in disease states.23 Matrix metalloproteinase 2 (MMP2) degrades type XI collagen by recognising Gly–Iso or Gly–Leu bonds24; however, Gly–Iso followed by Pro is resistant to collagenase.25 Increased activity of MMP2 destroys collagen architecture.26 Thus it is possible that threonine-621 in mutant collagen type XI α2 permits abnormal degradation of collagen type XI by MMP2 induced cleavage.
Conclusions
We report the first mutation in COL11A2 associated with ARNSHL (homozygosity for P621T in a consanguineous Iranian family). This missense mutation is close to the amino-terminus. Based on comparative genotype–phenotype studies of all COL11A2 mutations, which show that mutation type and location are important predictors of phenotype, and we hypothesise that P621T alters collagen folding or degradation.
Acknowledgments
This study was supported in part by a grant from the NIDCD (R01-DC02842) to RJHS and a grant from the Iran Deputy of Research and Technology, Ministry of Health and Medical Education (P 6176).
REFERENCES
Footnotes
-
Competing interests: none declared