Article Text

Abstract
Background: Subtelomeric rearrangements contribute to idiopathic mental retardation and human malformations, sometimes as distinct mental retardation syndromes. However, for most subtelomeric defects a characteristic clinical phenotype remains to be elucidated.
Objective: To screen for submicroscopic subtelomeric aberrations using multiplex ligation dependent probe amplification (MLPA).
Methods: 210 individuals with unexplained mental retardation were studied. A new set of subtelomeric probes, the SALSA P036 human telomere test kit, was used.
Results: A subtelomeric aberration was identified in 14 patients (6.7%) (10 deletions and four duplications). Five deletions were de novo; four were inherited from phenotypically normal parents, suggesting that these were polymorphisms. For one deletion, DNA samples of the parents were not available. Two de novo submicroscopic duplications were detected (dup 5qter, dup 12pter), while the other duplications (dup 18qter and dup 22qter) were inherited from phenotypically similarly affected parents. All clinically relevant aberrations (de novo or inherited from similarly affected parents) occurred in patients with a clinical score of ⩾3 using an established checklist for subtelomeric rearrangements. Testing of patients with a clinical score of ⩾3 increased the diagnostic yield twofold to 12.4%. Abnormalities with clinical relevance occurred in 6.3%, 5.1%, and 1.7% of mildly, moderately, and severely retarded patients, respectively, indicating that testing for subtelomeric aberrations among mildly retarded individuals is necessary.
Conclusions: The value of MLPA is confirmed. Subtelomeric screening can be offered to all mentally retarded patients, although clinical preselection increases the percentage of chromosomal aberrations detected. Duplications may be a more common cause of mental retardation than has been appreciated.
- ArrayCGH, array based comparative genomic hybridisation
- FISH, fluorescent in situ hybridisation
- MAPH, multiplex amplifiable probe hybridisation
- MLPA, multiplex ligation dependent probe amplification
- OFC, occipitofrontal head circumference
- mental retardation
- multiplex ligation dependent probe amplification
- polymorphism
- subtelomeric rearrangement
Statistics from Altmetric.com
- ArrayCGH, array based comparative genomic hybridisation
- FISH, fluorescent in situ hybridisation
- MAPH, multiplex amplifiable probe hybridisation
- MLPA, multiplex ligation dependent probe amplification
- OFC, occipitofrontal head circumference
- mental retardation
- multiplex ligation dependent probe amplification
- polymorphism
- subtelomeric rearrangement
Chromosomal rearrangements involving subtelomeric regions are a common cause of idiopathic mental retardation (reviewed by Knight and Flint1 and De Vries et al2). Subtelomeric rearrangements have been reported to occur in approximately 5% of patients with unexplained mental retardation.2 Some subtelomeric submicroscopic deletions result in well defined mental retardation syndromes, such as monosomy 1p36, Wolf–Hirschhorn syndrome (4p-), and cri-du-chat syndrome (5p-), but for most subtelomeric defects a characteristic phenotype remains to be defined.2 For this reason, screening of all subtelomeres is a valuable diagnostic tool. Multiprobe FISH (fluorescent in situ hybridisation), using telomeric probes on metaphase chromosomes is commonly used for detecting subtelomeric abnormalities.3–5 This reliable method, however, remains labour intensive and therefore expensive for routine diagnostic testing. New techniques, such as multiplex amplifiable probe hybridisation (MAPH) and array based comparative genomic hybridisation (arrayCGH), are proven to be suitable for the detection of subtelomeric chromosome aberrations.6,7 However, MAPH, requires immobilisation of sample nucleic acids8 and arrayCGH is labour intensive and requires expensive equipment. Recently, multiplex ligation dependent probe amplification (MLPA) was applied to detect subtelomeric defects in 75 patients with mental retardation of unknown cause.9 In the current study MLPA was used with a new set of subtelomeric probes designed by Schouten et al8 for the detection of submicroscopic aberrations in a larger sample of clinically well defined patients with idiopathic mental retardation. We show that MLPA is a reliable technique to detect submicroscopic telomeric copy number changes, rendering it suitable for routine diagnostic screening in mentally retarded patients.
METHODS
Patients
The diagnostic capacity of MLPA for detecting subtelomeric chromosome aberrations was tested by screening 210 patients with unexplained mental retardation. The patients were all referred to the department of human genetics, University Medical Centre Nijmegen for subtelomeric analysis. A total of 137 patients had been evaluated by one of the clinical geneticists at our centre. The remaining patients were referred by other medical specialists, mainly paediatricians. All patients had a normal G banded karyotype at a 550 band level using standard procedures, and no clinical syndrome had been recognised. The level of mental retardation (mild, IQ 50 to 70; moderate, IQ 30 to 50; severe, IQ <30) and the score on the checklist for submicroscopic subtelomeric rearrangements developed by de Vries et al10,11 were obtained retrospectively from notes by the referring specialist. The incidence of subtelomeric aberrations was assessed for the level of mental retardation and for the subtelomeric aberration checklist score. In case of a subtelomeric aberration, DNA samples of the parents were requested for further testing. The MLPA kit was first validated using 15 DNA samples from patients with 16 known (sub)microscopic defects involving the subtelomeric region.
Multiplex ligation dependent probe amplification
MLPA probes
A specifically designed set of probes for testing for subtelomeric chromosomal imbalances, SALSA P036 human telomere test kit (MRC-Holland, Amsterdam, Netherlands; http://www.mrc-holland.com) was used for subtelomere screening. Probe preparation has been described previously,8 The MLPA mix contained probes for each subtelomeric region (table 1) except for the short arms of the acrocentric chromosomes. For the latter, probe recognition sequences on the q arm, in one of the first genes following the repeated sequences of the centromere, were used. Because these probes were not subtelomeric, they were not included in our analysis.
Subtelomeric MLPA probes and their distance to the telomere
MLPA analysis
Genomic DNA of each patient was isolated using standard procedures.14 MLPA analysis was carried out as described by Schouten et al,8 with slight modifications. Briefly, 200 to 400 ng of DNA sample was diluted with milliQ to 8 µl and heated at 98°C for five minutes (GeneAmp PCR System 9700, Applied Biosystems, Foster City, California, USA). After addition of the probe mix (1.5 μl per sample), which was mixed 1:1 with a salt solution (1.5 M KCl; 300 mM Tris-HCl, pH 8.5; 1 mM EDTA), samples were heated for one minute at 95°C and incubated overnight at 60°C. Next the Ligation-65 mix (following the supplier’s instructions) was added and incubated for 15 minutes at 54°C. Ligase-65 was inactivated by heating at 98°C for one minute. The ligation products were amplified by polymerase chain reaction (PCR) using the common primer set with the 6-FAM label distributed by the supplier. Amplification products were identified and quantified by capillary electrophoresis on an ABI 3100 genetic analyser, using Genescan analysis software (version 3.7) and Genotyper software, all from Applied Biosystems. Subtelomeric screening of 48 samples using MLPA took approximately 1.5 days, including four hours of hands-on time. Each subtelomeric rearrangement was detected by at least one additional MLPA analysis.
Statistical analysis/data processing
The signal strength of the PCR products was determined by Genotyper software (Applied Biosystems). A spreadsheet was developed in Microsoft™ Excel in order to process the sample data efficiently. First, the data were normalised by dividing each probe’s signal strength by the average signal strength of the sample. This normalised peak pattern was divided by the average peak pattern of all the samples in the same experiment. The resulting values were approximately 1.0 for every wild type peak, 0.5 for heterozygous deletions, and 1.5 for heterozygous duplications. As a quality check for the probes, we computed the coefficient of variation (cv) of the normalised signal strength over the controls. If a particular probe had a cv of more than 10% over all samples tested, the results of the analysis for that particular probe were discarded. However, this was never the case with the Salsa P036 probe set. The analysis for a particular sample was repeated if the cv over all probes was more than 15% (∼18% of all tests). Twenty control samples (40 alleles) were run to exclude the presence of common polymorphisms and to test the feasibility of the statistical analysis.
Confirmation experiments
FISH analysis using first and second generation sets of telomere specific clones5,13 and Vysis probes (Vysis, Downes Grove, Illinois, USA) was carried out to confirm the aberrations identified by MLPA. Copy number changes were checked for de novo occurrence by MLPA and FISH analyses in both parents. In case the aberration could not be detected by FISH, DNA samples from the parents were only tested by MLPA. FISH analyses were done using routine methods. Fixed chromosome suspensions were prepared from cultured peripheral blood lymphocytes obtained from patients and parents. Labelling of the probes, slide preparation, and hybridisation were carried out using a standard protocol. A Leica DMRA fluorescence microscope, equipped with appropriate filters, was used for visual examination of the slides. The images were captured by a cooled CCD camera (SenSys) coupled to a Leica computer and analysed by a CW4000 software package. Inverted DAPI staining and a chromosome specific centromere probe were used for chromosome identification.
RESULTS
In this study MLPA was used to detect subtelomeric aberrations in a group of 210 patients with unexplained mental retardation. An improved set of subtelomeric probes—the SALSA P036 human telomere test kit—was applied. The sensitivity of the probe set was first determined by testing DNA samples from patients with a known chromosomal defect. These known defects were either cytogenetically visible or detected by FISH. Positive controls were available for 1pter, 1qter, 2qter, 3pter, 4pter, 7pter, 9pter, 10qter, 16qter, 18qter, 19qter, 22qter, and for the probes in the pseudoautosomal regions of Xpter, Ypter, Xqter, and Yqter. In all cases the genomic defect was confirmed by MLPA (data not shown).
The initial subtelomeric screening in patients with idiopathic mental retardation showed a subtelomeric rearrangement in 19 patients. Remarkably, a duplication of the 10qter MLPA probe was detected in five patients. These duplications could not be confirmed by FISH analysis. Parental analysis of two of the cases by the same technique revealed an identical duplication in a phenotypically normal father and mother in different families. In addition, the patients showed no clinical resemblance. For these reasons the 10qter duplication was considered most likely to be a polymorphism and was therefore not included in further analyses. Thus 14 patients (6.7%) with a subtelomeric rearrangement remained: 10 deletions and four duplications. Four MLPA profiles of subtelomeric aberrations are shown in fig 1. Five deletions were de novo (1p (twice), 3q, 4p, 10q) and all could be confirmed by FISH. Four deletions (2p, 11p, 12p, 16q) were also present in phenotypically normal parents and these deletions could not be confirmed by FISH. In case 10 with a 22qter deletion, the parents were not available for testing. Two de novo subtelomeric duplications (5q, 12p) were detected. The other duplications identified by MLPA (18q, 22q) were inherited from phenotypically similarly affected parents. For the 22qter duplication, FISH analysis showed that this was the result of a submicroscopic unbalanced translocation (t(21;22)), whereas the other direct duplications identified by MLPA could not be confirmed by FISH. Table 2 shows overviews of the respective submicroscopic deletions and duplications identified by MLPA.
Submicroscopic deletions and duplications identified by multiplex ligation dependent probe amplification (MLPA)
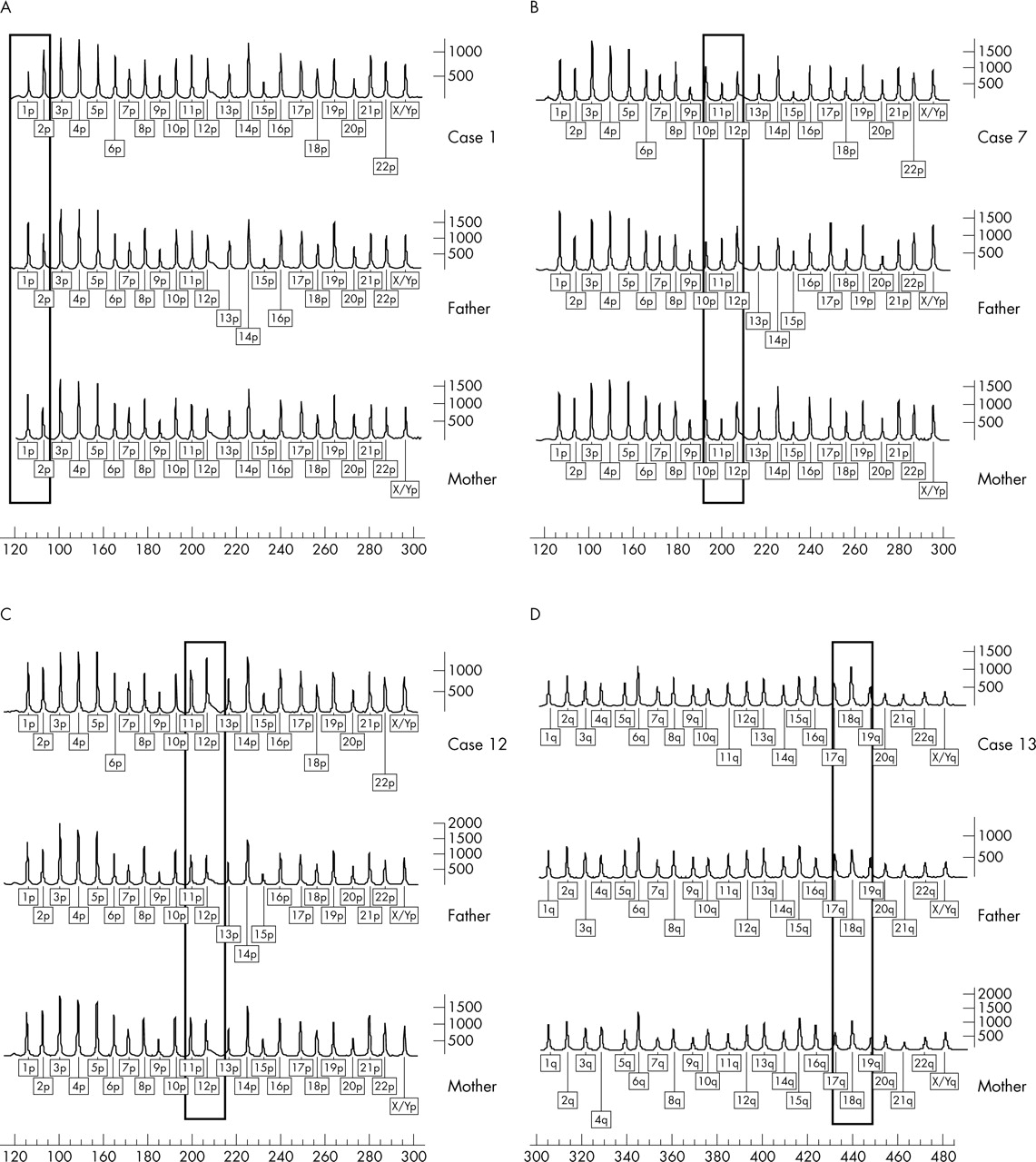
Detection of subtelomeric aberrations by multiplex ligation dependent probe amplification (MLPA). In each figure the profile of the patient is shown in line 1. The profiles of the father and mother are depicted in lines 2 and 3, respectively. The rectangle indicates the position of the aberrant MLPA probe. (A) De novo 1pter deletion (case 1). (B) Maternal inherited 11pter deletion (case 7). (C) De novo 12pter duplication (case 12). (D) 18qter duplication inherited from the mother (case 13).
The degree of mental retardation in the study group was mild in 31% of the cases (63/201), moderate in 39% (78/ 201), and severe in 30% (60/ 201). In nine cases the level of mental retardation was unspecified. Clinically relevant aberrations (de novo or inherited from a phenotypically similarly affected parent) occurred in 6.3%, 5.1%, and 1.7% of the mildly, moderately, and severely retarded patients, respectively. Figure 2 shows the clinical score on the checklist for subtelomeric rearrangements reported by de Vries et al10 and the subtelomeric anomalies per group. All aberrations with clinical relevance were identified in patients with a clinical score of ⩾3. Clinical information was insufficient for determining a score in 10 cases. A brief clinical description of the cases in which a chromosomal imbalance was identified is given below. If any phenotypic or chromosomal abnormality was present in either parent, this is included in the case description. For case 10 (del 22qter), a 14 year old boy, additional clinical features were not available, nor were the parents available for further testing.

{kind=link}
{kind=link}
Distribution of subtelomeric aberrations using the subtelomeric clinical checklist score developed by de Vries et al.10
De novo submicroscopic subtelomeric deletions identified by MLPA
Case 1 (del 1pter) was a moderately mentally retarded 18 month old girl (born at 40 weeks’ gestation; birth weight 3060 g, 15th centile). She presented with psychomotor delay and growth failure. She had a perimembranous ventricular septal defect, hearing loss, and several dysmorphic features including a large anterior fontanelle, prominent broad forehead, flat midface, deep set eyes, strabismus, downslant of the palpebral fissures, slight hypertelorism, small low set posteriorly rotated ears, small nose, and flat nasal bridge. In addition she had a flat palate with wide alveolar ridges, downturned corners of the mouth, pointed chin, a short neck, and small broad hands with abnormal implant of the thumbs. At the age of 11.5 months her height was 70 cm (5th centile) and her occipitofrontal head circumference (OFC) was 44.5 cm (16th centile).
Case 2 (del 1pter) was a severely mentally retarded five year old girl (born at 37 weeks’ gestation; birth weight 2765 g, 30th centile) who presented with delayed psychomotor development. Her dysmorphic features included almond shaped eyes, upward slant of the palpebral fissures, flat nasal bridge, bifid uvula, a small U shaped curve in the hard palate, bilateral epicanthic folds, downturned corners of the mouth, mild retrognathia, and a short perineum. She also had a urachus cyst which was corrected surgically and vesicoureteric reflux for which she was treated with prophylactic antibiotics. At four years and three months her height was 109.5 cm (75th centile), her weight was 28.6 kg (+4 SD), and her OFC was 49.9 cm (50th centile).
Case 3 (del 3qter) was a mildly mentally retarded six year old girl (born at 40 weeks’ gestation; birth weight 2930 g, 10th centile). She presented with hearing loss and psychomotor delay. She had scaphocephaly. Computed tomography of the cerebrum revealed a normal brain structure. At six years and two months her height was 1.17 cm (30th centile) and her OFC was 51 cm (50th centile). Besides the scaphocephaly, other dysmorphic features included frontal bossing, slightly downslanting palpebral fissures, low set posteriorly rotated ears, broad nostrils, smooth philtrum, everted lower lip, high palate, pectus carinatum, clinodactyly of the fifth fingers, long tapering fingers, and clinodactyly of toes three to five.
Case 4 (del 4pter) was a moderately mentally retarded five year old boy (born at 37+6 weeks’ gestation; birth weight 2105 g, −2.5 SD) who presented with psychomotor delay, growth failure, and severe feeding difficulties which required percutaneous endoscopic gastrostomy. He had unilateral hydronephrosis, astigmatism, and exotropia of the left eye. At the age of four years and 10 months his height was 97 cm (−3 SD) and his OFC was 46.2 cm (−3 SD). Dysmorphic features included plagiocephaly, frontal bossing, proptosis, epicanthus, hypertelorism, prominent glabella, wide nasal bridge, short philtrum, high palate, short neck, and hypospadias. In retrospect, the Wolf–Hirschhorn syndrome was the likely clinical diagnosis, although his psychomotor development was better than in previously reported cases and epilepsy was not present.
Case 5 (del 10qter) was a mildly mentally retarded two year old girl (born at 40+ weeks’ gestation; birth weight 2335 g, −2.5 SD). Delay in psychomotor development was noticed. At the age of 15 months her height was 70 cm (−3 SD) and her OFC was 44.5 cm (10th centile). Dysmorphic features included bilateral epicanthic folds, strabismus convergens (which was surgically corrected), posteriorly rotated left ear, and a small right ear with prominent helix. She underwent unilateral ureteric reimplantation because of left sided grade IV vesico-ureteric reflux with decreased function of the kidney. In addition she had a mild conductive hearing loss (40 dB) of the right ear.
Familial submicroscopic subtelomeric deletions identified by MLPA
Case 6 (del 2pter) was a moderately mentally retarded six year old boy (born at 40 weeks’ gestation; birth weight 3220 g, 20th centile). He presented with mild developmental delay, hyperactivity, and aggressive behaviour. Dysmorphic features included microcephaly, midface hypoplasia, bilateral epicanthic folds, small nose, smooth philtrum with a thin upper lip, short fifth fingers, and camptodactyly of the third left toe. Because of severe prenatal maternal alcohol abuse, fetal alcohol syndrome was suggested. At the age of five years and 10 months his height was 123 cm (85th centile) and his OFC was 48.5 cm (2nd centile).
Case 7 (del 11pter) was a moderately mentally retarded two year old girl born at full term (birth weight 2940 g, 30th centile). She presented with psychomotor delay and feeding difficulties. She had a triangular face, brachycephaly, deep set eyes, strabismus alternans, slight upslanting of the palpebral fissures, straight eyebrows, dysplastic helices, clinodactyly of the fifth digits of the hands, and hypoplastic nails of the second toes.
Case 8 (del 12pter) was a severely mentally retarded 10 month old boy (born at 35+6 weeks’ gestation; birth weight 2835 g, 50th centile). He was kept in hospital after delivery because of respiratory insufficiency and maternal fever during labour. He had microcephaly (−2.5 SD) and generalised hypotonia. Magnetic resonance imaging (MRI) of the cerebrum revealed semilobar holoprosencephaly with fusion of the frontal lobes and corpus callosum dysgenesis. Apart from a high palate no dysmorphic features were noted.
Case 9 (del 16qter) was a moderately mentally retarded nine year old boy born a term (birth weight 3800 g, 75th centile). He presented with psychomotor retardation with prominent speech delay. He had a triangular face, broad forehead, flat midface, slightly upslanting palpebral fissures, telecanthus, broad nasal bridge, large posteriorly rotated ears, and a prominent chin. He was admitted once to hospital because of haematuria of unknown cause. At nine years and two months his height was 132 cm (10th centile) and his OFC was 55 cm (86th centile).
De novo submicroscopic subtelomeric duplications identified by MLPA
Case 11 (dup 5qter) was a mildly mentally retarded three year old girl (41+4 weeks’ gestation) with bilateral mixed hearing loss (60 dB), epilepsy, a left sided choroidal defect, and a lateral neck fistula/dimple at the lower jaw. Psychomotor development was delayed, with initial walking at 26 months. MRI of the cerebrum showed enlargement of the intra- and extracerebral spaces. At the age of two years and 11 months her height was 95 cm (50th centile) and her OFC was 51 cm (85th centile). Dysmorphic features included brachycephaly, frontal bossing, midfacial dysplasia, narrow palate, small nose, small mouth, slight retrognathia, and small posteriorly rotated ears.
Case 12 (dup 12pter) was a moderately mentally retarded 12 year old girl (born at 37+4 weeks’ gestation; birth weight 2580 g, 30th centile) who presented with growth failure and hypermobility of the joints. She had feeding difficulties and constipation. At 12 years and four months of age her height was 141 cm (−2.5 SD) and her OFC was 51 cm (3rd centile). Dysmorphic features included high broad forehead, upslanting palpebral fissures, telecanthus, broad nasal bridge, short philtrum with a smooth upper lip, small maxilla, high palate, hypermobility of the wrists and the finger joints, and thin slightly hyperelastic skin. She had broad fingertips and bilateral clinodactyly of the fifth finger. In addition she had mild pectus excavatum and a strikingly furrowed tongue.
Familial submicroscopic subtelomeric duplications identified by MLPA
Case 13 (dup 18qter) was a mildly mentally retarded eight year old boy (39+5 weeks’ gestation; birth weight 2500 g, −2.5 SD). He was referred because of growth failure and mild mental retardation. At 7.5 years his height was 112.7 cm (−3.5 SD). Microcephaly was noted, with an OFC of 48 cm (−2.5 SD). His mother had a similar 18qter duplication and she had attended a special school for learning difficulties. Her adult height was 153 cm (−2.5 SD) with striking microcephaly (−4 SD).
Case 14 (dup 22qter) was a moderately mentally retarded five year old boy, born at term (birth weight 2330 g, 3rd centile) to unrelated parents. The first year of life was complicated by feeding problems, failure to thrive and frequent attacks of syncope. He presented at two years with delayed psychomotor development, seizures, hyperactivity, and dysmorphic features, including micro- and trigonocephaly, deep set eyes, flat midface, depressed nasal bridge, prominent upper lip, and downturned corners of the mouth. At the age of three years and 11 months his height and OFC were 93 cm (−3 SD) and 45.5 cm (−3.5 SD), respectively. MRI of the brain revealed post-haemorrhagic ventricular dilatation. Analysis of the father, who had similar clinical features (microcephaly, sparse hair, hypertelorism, and prominent upper lip), showed the same unbalanced submicroscopic translocation, der(21)t(21;22)(p10;q13.3), as was present in the boy.
DISCUSSION
In this study we present the results of the subtelomeric screening by MLPA in a group of 210 patients with unexplained mental retardation. A duplication of 10qter was identified in five of these patients on initial testing. This aberration was also present in two phenotypically normal parents and was regarded as a polymorphism and excluded from further analyses. Apart from the 10qter duplication, aberrations were identified in 6.7% of the patients screened: 10 deletions and four duplications. Clinically relevant submicroscopic aberrations were identified in nine patients (4.3%): seven de novo aberrations (five deletions and two duplications) and two duplications inherited from similarly affected parents. In the group of patients with mild, moderate, and severe mental retardation, clinically relevant anomalies occurred in 6.3%, 5.1%, and 1.7% of the cases, respectively.
It is of note that the greatest frequency of abnormalities was detected among mildly mentally retarded patients, in contrast to previous findings by Knight et al, who reported abnormalities in 7.4% among moderately or severely retarded individuals, and only in 0.5% among mildly retarded individuals.3 This might be explained by the increased detection of smaller aberrations and by the identification of submicroscopic duplications that cause less severe phenotypes in general. Our data support testing for subtelomeric aberrations in individuals with mild mental retardation.
In addition, rearrangements with clinical relevance were all found in patients with a clinical score of ⩾3 using the checklist for subtelomeric rearrangements composed by de Vries et al.10 This 0–10 checklist was developed to help preselection of cases for subtelomeric testing and consists of five items: family history of mental retardation, prenatal onset of growth retardation, postnatal growth abnormalities, two or more facial dysmorphic features, and one or more non-facial dysmorphic features or congenital abnormalities. Testing of patients with a clinical score of ⩾3 increased the diagnostic yield twofold to 12.4% (12/97).
The results of the current study show that clinical preselection of cases for subtelomeric screening is beneficial. The frequency of abnormalities in this study is comparable to previous studies, in which subtelomeric defects were identified in approximately 5% of the patients,2,16 although direct telomeric duplications were not included in the previous studies. A novel finding in our study is that subtelomeric direct duplications are a relatively frequent cause of isolated as well as familial mental retardation. Previous studies using FISH strategies were largely insensitive to such duplications, except those associated with an unbalanced translocation. We believe that subtelomeric direct duplications may have been underdiagnosed.
We identified five de novo submicroscopic subtelomeric deletions in this study. del 1pter (case 1–2), del 4pter (case 4), and del 10qter (case 5) have often been reported before and resembled the previous reports.2 The patient with a de novo 3qter deletion (case 3) had scaphocephaly, which was also seen in her mother (OFC 60 cm; +2.5 SD), grandfather, two of uncles, and a cousin. A de novo submicroscopic 3qter deletion has only been reported once.17 In microscopically visible terminal deletions of 3q, similar abnormal skull shapes (dolichocephaly and trichonocephaly) have also been reported.18 However, the abnormal skull shape seemed to be familial and therefore not to be related to the de novo 3qter deletion in our case.
Two de novo submicroscopic subtelomeric duplications were identified. The three year old mildly mentally retarded girl with a duplication 5qter (case 11) had to our knowledge the first reported submicroscopic duplication of this region. In addition to facial dysmorphisms, she had hearing loss and epilepsy. Patients with microscopically visible 5qter duplications and unbalanced 5qter duplications with additional deletions of other chromosome ends have been described.19,20 In addition to mental retardation, growth retardation, seizures, and some overlapping facial characteristics were present, making it likely to be a pathogenic cause in this patient. The 12pter duplication is also the first submicroscopic duplication of this region to be reported. Some of the dysmorphisms found in microscopically visible cases of 12pter duplications21 could also be observed in our case.
Four subtelomeric deletions were inherited from phenotypically normal parents. The phenotypes described in these cases—del 2pter (case 6), del 11pter (case 7), del 12pter (case 8), and del 16qter (case 9)—were quite different from previously reported cases with deletions in a similar region.17,22–25. Therefore it is likely that these aberrations are polymorphisms without clinical implications.
Both familial duplications detected in this study were inherited from similarly affected parents. In case 13, a maternal duplication of probe 18qter was identified in a mildly retarded boy presenting with growth failure, and the same features were observed in his mother. She was microcephalic (−4 SD) and had a height of −2.5 SD and learning difficulties. Intrauterine growth retardation and microcephaly have been reported in microscopically visible 18qter duplications,26 strengthening the impression that the duplication in the 18qter chromosomal segment caused the phenotype in our patient. The duplication in our patient was significantly smaller than those previously reported, which might explain the milder phenotype in our case. In case 14, MLPA analysis showed a duplication of the 22qter probe. FISH analysis confirmed the duplication and revealed an unbalanced submicroscopic translocation—der(21)t(21;22) (p10;q13.3)—in the proband and his father. The father was mildly mentally retarded and showed similar minor facial anomalies. Duplications including the telomere region of the long arm of chromosome 22 have been described, with some clinical resemblance to the proband, such as intrauterine growth retardation, microcephaly, and hypertelorism.27
All de novo deletions could be confirmed by FISH analysis. However, the inherited deletions (del 2qter, del 11pter, del 12pter, and del 16qter) could not be confirmed. The MLPA probes were positioned in or close to (∼50 kb) the telomere specific FISH clones of the same telomere, except for the 11pter MLPA probe which was mapped more to the centromere (UCSC Genome Browser, July 2003 Freeze).5 It is most likely that these familial aberrations represent small genomic polymorphisms missed by a FISH probe encompassing the same region. Three duplications (dup 5qter, dup 12pter, and dup 18qter) could not be confirmed by FISH. The duplicated regions were probably too close together on the genome to be detectable by routine FISH.
In the current study a single new set of subtelomeric probes was used—the SALSA P036 human telomere test kit. In general the MLPA probes were either located in the region covered by the telomere specific FISH clone or were closely proximal to this region. For the latter probes, it is possible that terminal aberrations detectable by FISH may be missed by MLPA. In a recent paper, a group of 75 mentally retarded patients was analysed using two complementary MLPA probe sets, SALSA P019 and P020.9 When validating these probe sets on our panel with known chromosomal aberrations, we found that several probes in the P019 and P020 kit were too far from the telomere to detect small terminal deletions. The main problem is that the probe on 1p missed small deletions that are relatively common.28 Furthermore, in addition to the probe on 21q,9 the probes for 2q, 6p, and 15q in the P019 and P020 kit were polymorphic (data not shown). Thus the P036 kit not only has the advantage that only one kit is needed for all subtelomeric regions, but the individual probes in this kit are also more reliable. A fusion of the three kits with additional subtelomeric probes might eventually offer the best solution for routine diagnostic screening of subtelomeric aberrations because it will allow more accurate identification and delineation of the subtelomeric copy number changes.
Conclusions
We have confirmed that MLPA is a reliable method for detecting subtelomeric rearrangements. Screening for subtelomeric anomalies by MLPA can be offered to all mentally retarded patients, although clinical preselection increases the percentage of anomalies detected. To exclude polymorphisms, interpretation of the results should always include parental testing and comparison with clinical features of previously reported patients with similar subtelomeric rearrangements. In addition, we found that MLPA detects pure subtelomeric duplications that can easily be missed by routine FISH analysis.
Note added in proof
The duplication 5qter in case 11 could not be confirmed by later MLPA testing when repeated on a new DNA sample and therefore we cannot exclude the possibility that the duplication was the result of a technical artefact. Since the acceptance of this paper, we have found several other duplications in the same subtelometric region—for example on 15qter and 9pter—confirming that duplications may be more common than previously thought.
Acknowledgments
We thank all the patients and their families for their kind cooperation. Bert de Vries was supported by a grant from ZON-MW (the Netherlands).
REFERENCES
Footnotes
-
Conflicts of interest: none declared