Article Text

Abstract
Starting from a cohort of 50 NADH-oxidoreductase (complex I) deficient patients, we carried out the systematic sequence analysis of all mitochondrially encoded complex I subunits (ND1 to ND6 and ND4L) in affected tissues. This approach yielded the unexpectedly high rate of 20% mutation identification in our series. Recurrent heteroplasmic mutations included two hitherto unreported (T10158C and T14487C) and three previously reported mutations (T10191C, T12706C and A13514G) in children with Leigh or Leigh-like encephalopathy. The recurrent mutations consistently involved T→C transitions (p<10−4). This study supports the view that an efficient molecular screening should be based on an accurate identification of respiratory chain enzyme deficiency.
- CT, computed tomography
- MRI, magnetic resonance imaging
- OFC, occipital frontal circumference
Statistics from Altmetric.com
The twofold genetic origin of the mitochondrial respiratory chain, the large number of catalytic and assembly proteins, and the allelic/non-allelic heterogeneity of disease causing mutations make identification of the mutant genotypes in respiratory chain deficiency particularly tedious. However, elucidating the disease mechanism is of particular importance for establishing the mode of inheritance and identifying the recurrence risk in affected families.
Starting from a cohort of 50 NADH-oxidoreductase (complex I) deficient patients, we carried out the systematic sequence analysis of all mitochondrially encoded complex I subunits in affected tissues. This systematic approach yielded the unexpected mutation rate of 20% including recurrent de novo mtDNA mutations in our series. Whatever the mechanism, this high rate of recurrent de novo mutations is important for genetic counselling. In addition, this study gives support to the view that an efficient molecular screening should be based on accurate identification of the respiratory chain deficiency causing the disease.
PATIENTS
Patient 1
A girl was born to unrelated healthy parents after a 38 week pregnancy (weight 3320 g, length 48.5 cm, occipital frontal circumference (OFC) 34 cm). Her elder sister is healthy. Psychomotor development was apparently normal until 4 months of age when poor visual contact, floppiness, and abnormal movements were noted. Elevated plasma (8 mmol/l) and CSF lactate were noted (7 mmol/l, control below 2.2 mmol/l). Magnetic resonance imaging (MRI) showed widespread signal anomalies consistent with the diagnosis of Leigh disease, namely bilateral involvement of the putamen and of the lateral dorsal and dorsomedial thalamic nuclei, pulvinar, substantia nigra, and external globi pallidi, associated with involvement of the spinothalamic tracts and medial lemniscus, and of the reticular formation. A cortico-subcortical hypersignal was also seen in the cingular gyri and in the central regions. MR spectroscopy detected a very high peak of lactate in the basal ganglia. The patient died at 5 months of age after a rapid neurological deterioration.
Patient 2
A boy was born to unrelated healthy parents after a 37 week pregnancy (weight 2600 g, length 47 cm, OFC 33 cm). His elder sister is healthy. He was first admitted aged 19 days for jaundice. At 4 weeks, persistent failure to thrive with high plasma (5.4–7.5 mmol/l) and urinary lactate, and high plasma lactate/pyruvate molar ratio were noted (27–37, normal <15). Brain MRI at 6 weeks of age showed a high T2 signal intensity in the putamen suggestive of Leigh’s encephalopathy. MR spectroscopy detected a peak of lactate in the right lenticular nucleus. Heart ultrasound and fundus were normal. The patient died at 7 months after a rapid neurological deterioration.
Patient 3
A boy was born to unrelated healthy parents after a 39 week pregnancy (weight 3700 g, length 49 cm, OFC 34 cm). Two maternal aunts and two maternal uncles had died at 3–16 months of age of an unexplained encephalopathy. He developed normally until 15 months of age when he presented acute ataxia, drowsiness, and vomiting without lactic acidosis. Brain NMR at the age of 2 years showed bilateral involvement of internal globi pallidi, middle part of the putamen, and the caudal part of the caudate nuclei, suggestive of Leigh’s encephalopathy. Pons and medulla were normal. At 32 months, he suddenly developed a second attack of regression with dystonia and dysarthria. His psychomotor development slowly improved thereafter but he is wheelchair bound with severe spaticity, residual dystonia, and facial dyskinesia.
Patient 4
A girl was born to unrelated healthy parents after a 38 week pregnancy. She was small for age (weight 2400 g, length 46 cm, OFC 32 cm). Trunk hypotonia was present at birth. She failed to thrive and developmental milestones were delayed. She could not follow movement with eyes, and developed myoclonic jerks and pyramidal syndrome. Her plasma lactate was elevated (8 mmol/l). Brain MRI showed signal intensity in the putamen suggestive of Leigh’s encephalopathy.
Patient 5
A boy was born to unrelated healthy parents after a term pregnancy. His elder sibling is healthy. He gradually developed psychomotor retardation of unexplained origin and suddenly presented an attack of regression, drowsiness, vomiting, myoclonic jerks, massive hypotonia, hypothermia, bradypnoea, and bradycardia at 26 months of life. His plasma lactate was elevated (6 mmol/l). A computed tomography (CT) scan performed at 18 months of age was normal, but CT scan at 3 years showed an acute bilateral involvement of the putamen and globi pallidi, and a severe cortico-subcortical atrophy. At 9 years, brain MRI showed hypersignal in the bulbar nuclei, suggestive of Leigh’s encephalopathy and supratentorial anomalies. His psychomotor development slowly improved thereafter but at 12 years, he is severely disabled, has no speech and is wheelchair bound.
Patient 6
A girl was born to unrelated healthy parents after a normal pregnancy (weight 3 kg, length 48 cm, OFC 34.5 cm). Her elder sister and brother are healthy. She did well during the first year of life. At that age, eyelid ptosis with fatigue and mild motor delay were noted. At 5 years, the bilateral ptosis persisted with ophthalmoplegia. Plasma lactate was elevated (5 mmol/l). Brain MRI showed a lacuna in the cerebral pedoncula. She has normal schooling at 16 years of age.
Patient 7
A boy born to unrelated parents presented a generalised epilepsy ascribed to astrocytoma at the age of 27 years after an uneventful childhood and adolescence. At 33 years of age, he developed proteinuria and haematuria leading to the diagnosis of tubulointerstitial nephropathy, associated with hypertrophic cardiomyopathy. At this age, peripheral neuropathy, optic atrophy, and mild psychomotor retardation were noted. The patient developed diabetes mellitus at 35 years of age but no migraine, stroke-like episodes, or lactic acidosis was noted.
MATERIALS AND METHODS
Polarographic and spectrophotometric studies of respiratory chain enzymes were carried out on homogenates and mitochondria enriched fractions from scaled down skeletal muscle biopsies (100 mg, deltoid) and on endomyocardial biopsies of the children.1 NADH-ubiquinone oxidoreductase (complex I) activity was measured as described.1 For chorionic villi, complex I activity was measured as the rotenone sensitive decylubiquinone reductase activity after treatment of the cells in 1 ml ice cold solution consisting of 0.25 mol/l sucrose, 20 mmol/l Tris (pH 7.2), 40 mmol/l KCl, 2 mmol/l EGTA and 1 mg/ml bovine serum albumin, 0.01% digitonin (w/v) and 10% Percoll (v/v) as recently described.2 Because a balanced proportion between complexes is required for normal functioning of the respiratory chain, enzyme activities were presented both as absolute values and as ratios. For molecular studies, genomic DNA was extracted from skeletal muscle, blood leukocytes, skin fibroblasts, or liver by standard methods. Mitochondrial DNA subunits of complex I (ND1 to 6 and ND4L) were submitted to PCR amplification after an initial denaturation at 96°C for 5 minutes, followed by 20 cycles of 96°C for 30 seconds, 55°C for 30 seconds and 72°C for 30 seconds, and a last extension at 72°C for 10 minutes. Cross hybridisation of oligonucleotide primers to genomic DNA was ruled out using mtDNA-less Rho° cells3 in each PCR reaction. PCR products were electrophoresed onto a 2% agarose gel, purified by ExoSapIT® (Amersham Pharmacia Biotech, Amersham, Buckinghamshire, UK) and directly sequenced using the PrismTM Ready Reaction Sequencing Kit (PE Applied Biosystems, Foster City, CA, USA) on an automatic sequencer (ABI 3100; PE Applied Biosystems). The sequences were then analysed by the SeqScape™ software (PE Applied Biosystems).
The screening of the T10158C mutation was carried out using the allele creating restriction site method. The forward primer spanned nucleotides 10131–10157 with two mismatches (CC) at 10155–10156, creating a BamHI site at 10158. The reverse primer extended from 10329 to 10350. BamHI digestion of the wild type amplification product generated two fragments of 196 bp and 24 bp respectively, while mutant DNA was undigested (220 bp). The level of heteroplasmy was estimated by scanning the wild type and mutant amplification products. The screening of T10191C and T12706C mutations was carried out according to Taylor et al.4,5 The screening of the A13514G mutation was carried out as described for the G13513A mutation.6
RESULTS
Polarographic and spectrophotometric evidence of complex I deficiency was found in skeletal muscle biopsy of a series of 50 patients. The scattering of control values occasionally hampered the recognition of enzyme deficiencies, as normal values frequently overlapped those found in patients. For this reason, the results were expressed as ratios, especially as the normal functioning of the respiratory chain requires a constant ratio of enzyme activities. Under these conditions, patients whose absolute activities were in the low normal range could be unambiguously diagnosed as enzyme deficient.1 Neither large scale mtDNA rearrangements nor common mtDNA mutations were found in their muscle. Thus, the systematic sequence analysis of the seven mitochondrially encoded complex I subunits (ND1–ND6 and ND4L)7,8 and DHPLC sequence analysis of nuclear encoded complex I cDNAs (NDUFS1, NDUFS2, NDUFS4, NDUFS7, NDUFS8, NDUFV1) were performed in the 50 patients. Seven of them were found to carry mutations in mitochondrially encoded subunits of complex I (table 1).
Enzymological studies
Patients 1 and 2, who are unrelated, carried the same hitherto unreported heteroplasmic T→C transition at nucleotide 10158 in the mitochondrially encoded ND3 gene (T10158C) changing a serine into a proline in the protein (S34P, fig 1). A high proportion of mutant molecules was found in their skeletal muscle (heteroplasmy = 85% of mutant molecules) but not in the blood leukocytes of the parents, suggesting a de novo mutation. The T10158C mutation was found in the background of the European haplogroup U in patient 2 but in the background of a different unreported haplogroup in patient 1. A prenatal diagnosis was offered to the parents of patient 1 for the next pregnancy. Complex I activities on trophoblast and amniotic cells were in the normal range (table 1) and molecular studies failed to detect the T10158C mutation in trophoblast and amniotic cells of the foetus. A boy was born and was healthy when examined at 5.5 months of age.
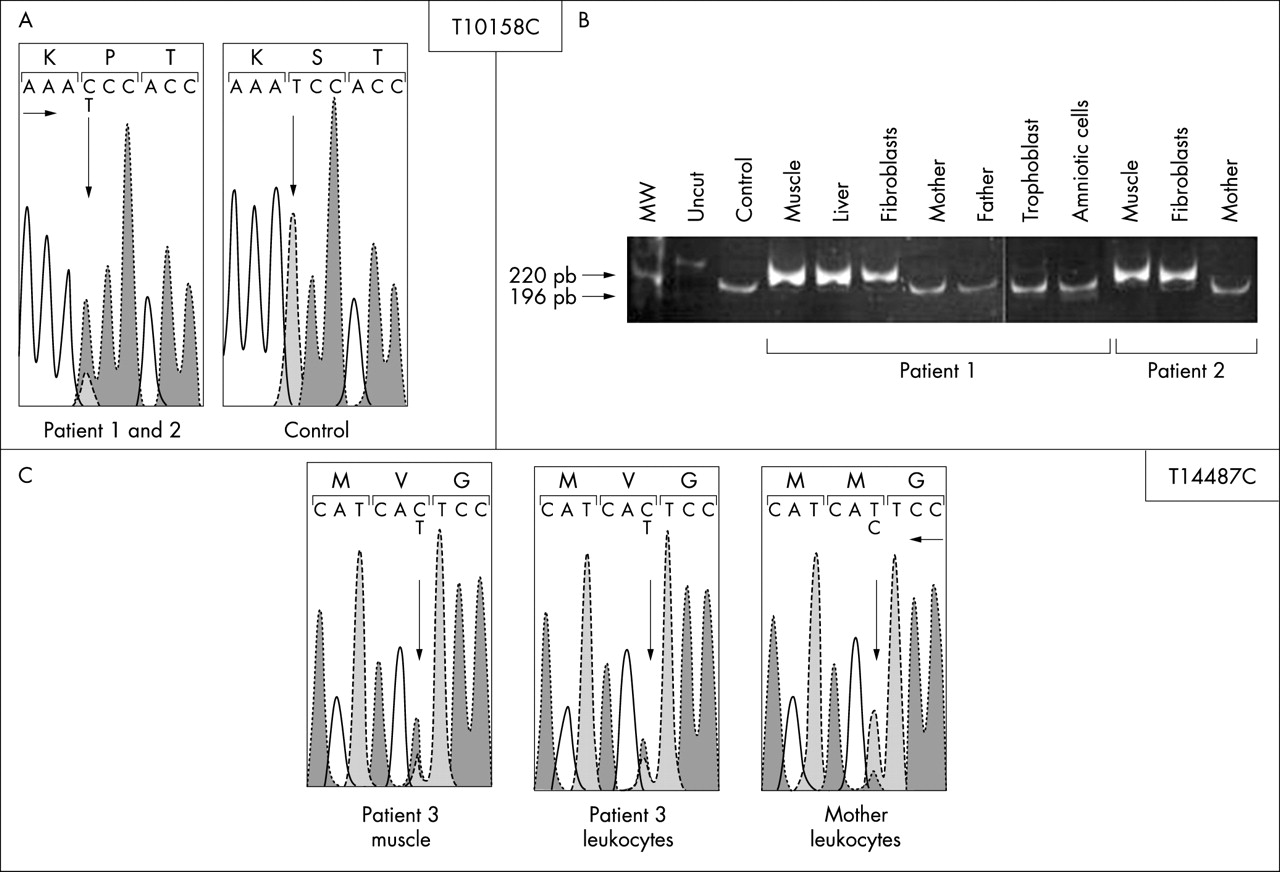
{kind=link}
Molecular genetic analysis of the T10158C and T14487C mutation. (A) Sequence of the ND3 gene showing the heteroplasmic change in patients 1 and 2 compared with control. (B) Quantification of the relative amount of wild type (196 bp) and mutant (220 bp) mtDNA in the family members of patients 1 and 2. (C) Sequence of the ND5 gene showing the heteroplasmic change in patient 3 compared with a control.
In skeletal muscle of patient 3, a previously unreported heteroplasmic T→C transition at nt 14487 of the mitochondrially encoded ND6 gene (T14487C, heteroplasmy = 80% of mutant molecules) changed a methionine into a valine in the protein (M63V, fig 1). A low level of mutation was detected in leukocytes of his mother (≈15%, fig 1).
Previously reported mutations were found in muscle or heart of patient 4–7, namely the ND3 T10191C4 (patients 4–5, heteroplasmy = 90 and 80% of mutant molecules), ND5 T12706C5 (patient 6, heteroplasmy = 60% of mutant molecules) and ND5 A13514G mutation9 (patient 7, heteroplasmy = 90% of mutant molecules), but none of their parents was found to carry these mutations in their circulating leukocytes.
The five mutations reported here were recurrent mutations in unrelated families. They were randomly distributed along the mtDNA circle and were not found in 560 mtDNA genomes sequenced by the MITOKOR Corporation,10 supporting their relevance in the disease. They were not clustered in any specific region known to be particularly exposed during mtDNA replication. It is worth noting that these mutations consistently involved thymine to cytosine transitions within TCC or TTCC motifs, either on the coding strand (T10158C, T10191C, T12706C, T14487C) or the non-coding strand of the mt DNA molecule (A13514G). A total of 65 disease causing base substitutions in proteins coding genes have been reported (http://www.mitomap.org, last edited 1 April 2003). Instead of a balanced repartition of the 12 expected transition/transversion mutations, a marked skewing toward T→C (25%) and G→A transitions was noted (41.5%). This difference is highly significant (T→C transition p<10−4, G→A transition p<10−4) but remains hitherto unexplained.
DISCUSSION
Here we report on recurrent de novo or inherited mtDNA mutations in unrelated patients with Leigh or Leigh-like encephalopathy and NADH oxidoreductase (complex I) deficiency in their skeletal muscle biopsy. Heteroplasmic mutant genotypes included three previously reported (T10191C, T12706C, A13514G) and two hitherto unreported mutations (T10158C, T14487C). The T14487C mutation occurred in a region of the ND6 gene that is highly conserved across species. The T10158C mutation involved a less conserved region of the ND3 gene. The heteroplasmy of these mutations in the probands, their absence from circulating leukocytes of the patients’ mothers and from controls, and the conservation of the mutated amino acids across species strongly support the idea that these mutations cause the disease.
The random meiotic (patient 3) or mitotic partitioning of mtDNA mutations (patients 1, 2, and 4–5) probably accounts for the variability of clinical presentations among individuals carrying the same mutation. Indeed, the T10191C mutation causing Leigh or Leigh-like syndrome in patients 4–5 was first reported for epilepsy strokes optic atrophy cognitive decline syndrome.4 The T12706C mutation detected in patient 6 has already been reported for Leigh syndrome.5 Similarly, the A13514G mutation detected in patient 7 was first reported in two patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS).9 Alternatively, it is conceivable that other genetic factors (including nuclear factors) could play a role in the partitioning of mtDNA mutations.
If five recurrent mtDNA mutations reported here in 7/50 unrelated complex I deficient patients are compared with the recurrent G13513A mutation reported by Chol et al in 3/50 unrelated complex I deficient children of the same cohort,6 it appears that the systematic screening of all mitochondrially encoded complex I subunits resulted in an unexpectedly high mutation detection rate in our series (20% of our complex I deficient children). This unexpectedly high rate is of particular importance for genetic counselling.
Why specific mtDNA residues were recurrently targeted in respiratory chain deficiency remains unexplained. Why the recurrent mutations reported here consistently involved T→C transitions on either the coding (4/5) or the non-coding strand of the double helix (1/5) is also questionable. Consistently, it is worth noting that the Mitomap database of reported human mitochondrial DNA base substitution diseases contains a majority of G→A (41.5%) or T→C transitions (25%).
In conclusion, the present report shows that the systematic sequence analysis of mtDNA encoded complex I subunits yielded an unexpectedly high rate of mutations in our series. This study supports the view that an efficient molecular screening of respiratory enzyme deficiency should be based on the accurate identification of the deficiency causing the disease.
Acknowledgments
We are thankful to Professor M Radman for his interesting discussion and commentaries. We thank the Association Française contre les Myopathies for support.