Article Text

Abstract
Germline mutations in the BHD gene cause the dominantly inherited cancer susceptibility disorder, Birt–Hogg–Dubé (BHD) syndrome. Individuals with BHD are reported to have an increased risk of renal cell carcinoma (RCC) and of colorectal polyps and cancer. The BHD gene maps to 17p11.2, and to investigate whether somatic inactivation of the BHD gene region is implicated in the pathogenesis of sporadic RCC and colorectal cancer (CRC), we performed mutation analysis in 30 RCC primary tumours and cell lines, and 35 CRCs and cell lines. A somatic missense mutation (Ala444Ser) with loss of the wild type allele (consistent with a two hit mechanism of tumorigenesis) was detected in a primary clear cell RCC, and a further missense mutation (Ala238Val) was identified in a clear cell RCC cell line for which matched normal DNA was not available. A somatic missense substitution (Arg392Gly) was identified in a primary CRC, and the same change was detected in three RCCs (all oncocytomas) for which matched normal DNA was not available. A germline Arg320Gln missense variant detected in a primary CRC was not detected in 40 control individuals or in a further 159 familial and sporadic CRC cases. However, AA homozygotes for an intronic single nucleotide polymorphism (c.1517+6 G→A) were under-represented in familial cases compared with controls (p = 0.03). For some tumour suppressor genes, epigenetic silencing is a more common mechanism of inactivation than somatic mutations. However, we did not detect evidence of epigenetic silencing of BHD in 19 CRC and RCC cell lines, and BHD promoter region hypermethylation was not detected in 20 primary RCCs. These findings suggest that BHD inactivation occurs in a subset of clear cell RCC and CRC.
- BHD
- cancer
- colorectal
- renal
- BHD, Birt–Hogg–Dubé
- CRC, colorectal cancer
- MSI, microsatellite instability
- PCR, polymerase chain reaction
- RCC, renal cell carcinoma
- SSCP, single strand conformational polymorphism
- TSGs, umour suppressor genes
- VHL, von Hippel-Lindau disease
Statistics from Altmetric.com
- BHD, Birt–Hogg–Dubé
- CRC, colorectal cancer
- MSI, microsatellite instability
- PCR, polymerase chain reaction
- RCC, renal cell carcinoma
- SSCP, single strand conformational polymorphism
- TSGs, umour suppressor genes
- VHL, von Hippel-Lindau disease
The identification of rare familial cancer genes has provided important insights into the pathogenesis of both familial and sporadic cancers. Thus, the isolation of the susceptibility genes for von Hippel-Lindau (VHL) disease and familial adenomatous polyposis coli led to the demonstration that somatic inactivation of the VHL and APC tumour suppressor genes (TSGs) has a critical role in the development of most sporadic clear cell renal cell carcinoma (RCC) and colorectal cancer (CRC) respectively.1–4
Birt–Hogg–Dubé (BHD) syndrome is a multisystem, dominantly inherited, familial cancer syndrome characterised by the development of benign skin tumours on the face and upper body in the third decade of life.5 Histological examination of the tumours reveals them as fibrofolliculomas or trichodiscomas. Additional features include susceptibility to pneumothorax and RCC,10,11 and in some reports, colorectal polyps and CRC.6,7 Whereas RCC in VHL disease is always of a clear cell (conventional) type, a variety of histopathological subtypes have been described in BHD.11 The BHD gene maps to 17p11.2, and an inherited RCC syndrome in German Shepherd (Alsatian) dogs maps to the orthologous region of the canine genome.12–14 The BHD gene was identified recently and encodes a 64 kDa protein, folliculin, of unknown function.15 To date, germline BHD mutations have been described in 11 kindreds, and a mutation hotspot at a mononucleotide tract (C8) in exon 11 has been identified.15,16
Somatic inactivation of the VHL TSG occurs as an early event in ∼60% of clear cell RCCs.2–3,17 To date, mutation analysis of pVHL interacting proteins have not revealed mutations in sporadic clear cell RCC without VHL inactivation. In order to evaluate the potential role of the BHD TSG in the pathogenesis of sporadic human cancers, we analysed RCC tumours and cell lines (including primary RCC without VHL inactivation), and in sporadic CRCs and cell lines for somatic mutations. In addition, as epigenetic silencing by promoter region hypermethylation is a major mechanism of TSG inactivation in many cancers,18 we also looked for evidence of epigenetic inactivation of BHD in RCC and CRC.
MATERIALS AND METHODS
Patients and samples
We focused on two types of cancer: CRC and RCC. For BHD mutation analysis we initially analysed six colorectal cell lines (SW48, LS 174T, LoVo, LS411, HCT 116, and DLD-1) and nine kidney cell lines (SKRC18, SKRC39, SKRC45, SKRC47, SKRC54, KTCL26, 786-O, UMRC-2, and UMRC-3). Tumour samples analysed in this study were mostly of sporadic origin. In total, 50 tumour samples were analysed, of which 29 were colorectal tumours and 21 were RCCs (six oncocytomas and 15 clear cell RCC tumours with no known VHL mutations). Some information on the tumours has been published previously.19,20 Three samples were associated with ulcerative colitis, an additional two patients had multiple colorectal adenomas, one case had familial CRC and two cases had synchronous cancers. DNA was extracted from tumour and normal tissue (normal mucosa for CRC and blood or matched kidney) by standard methods. After informed consent, germline DNA (blood) from an additional 159 cases of familial and sporadic CRC were analysed for exon 9 variants. Laboratory controls matched for age and ethnic group were used for allele frequency comparisons.
BHD mutation analysis
BHD mutation analysis was performed as described in Nickerson et al.15 In brief, 14 exons were amplified by polymerase chain reaction (PCR). Amplicons were obtained from a PCR reaction of 50–100 ng of genomic DNA, 20 pmol/l of each primer, 0.25 mmol/l dNTPs, 1.5 mmol/l MgCl2 and 0.8 U of Taq polymerase enzyme (Sigma Chemical Co., Poole, Dorset, UK).
Single strand conformational polymorphism (SSCP) analysis was performed using standard methods.21 Samples presenting an abnormal pattern were then selected for further analysis (fig 1). Four exons (7, 9, 11 and 12) in which BHD mutations have been reported previously were sequenced in all cases, and 10 samples with negative SSCP analysis were also sequenced for each of the remaining exons, but no intragenic sequence variants were detected that had not already been detected by SSCP. Microsatellite instability was assessed with five microsatellite markers: D2S123, D8S255, D13S175, BAT26, and BAT40.

BHD mutation analysis (by SSCP and confirmed by sequencing). (A) Germline mutation found in exon 9. (B, C) Somatic mutations in exons 10 and 12 respectively.
Sequencing of PCR products
PCR products were cleaned up by an enzymatic treatment using exonuclease I to digest excess PCR primers, and shrimp alkaline phosphatase (both Amersham Biosciences, Amersham, Bucks, UK) to degrade excess nucleotides carried over from the PCR. The treatment was carried out for 15 minutes at 37°C and denatured for further 15 minutes at 80°C. Products were confirmed by direct sequencing from the forward and reverse PCR primers using the ABI Prism® BigDye™ Terminator cycle sequencing kit according to the manufacturer’s instructions and analysed on an ABI 377 DNA Analyzer (Perkin Elmer, Beckinsfield, Bucks, UK).
BHD promoter methylation analysis
Cell lines and patients’ samples
A total of 10 colorectal (SW48, LS 174T, LoVo, LS411, HCT 116, DLD-1, HAC7, SW480, SW620, HT-29) and nine kidney (SKRC18, SKRC39, SKRC45, SKRC47, SKRC54, KTLC26, 786-0, UMRC2, UMRC3) tumour cell lines were analysed. Twenty primary kidney tumours and 10 corresponding normal kidney DNA samples were also analysed.
5-aza-2′-deoxycytidine treatment of cell lines
5-aza-2′-deoxycytidine (5-aza-dC; Sigma) was freshly prepared in double distilled H2O (at 2mg/ml) and filter sterilised. In a 75 cm2 flask, 1×106 cells were plated in RPMI 1640 media supplemented with 10% fetal calf serum and left to settle for 24 h (day 0). Colorectal and kidney cell lines were treated with 2 μmol/l 5-aza-dC for 5 days. Both tumour cell lines were treated with 5-aza-dC on days 1, 3, and 5 and harvested on day 6, with a change of medium on days 2 and 4.19,20
BHD gene expression analysis by RT-PCR
RNA was prepared after treatment using RNABee (AMS Biotechnologies, Milton, Oxfordshire, UK) according the manufacturer’s guidelines.
BHD gene expression was determined by RT-PCR using two pairs of primers: 5′-ATG AACAGTCGGATGCGTG-3′ and 5′-TGGTGTAGGAATGGCGTGA-3′, producing an amplicon of 536 bp, and 5′-GGAGTGGATGAACAAAGTGA-3′and 5′-AACCTCGGGAGCAGACAT-3, resulting in a PCR product of 595 bp.
GAPDH was used as positive control, where the GAPDH primers were (5′-GACCCCTTCATGACCTCAACTACA-3′ and 5′-CTAAGCAGTTGGTGGTGCAGGA-3′), producing a PCR product of 369 bp.
Bisulphite modification and methylation analysis
Bisulphite DNA sequencing was performed as described previously.19,20 Briefly, 0.5–1.0 μg of genomic DNA was denatured in 0.3 mol/l NaOH for 15 min at 37°C. Unmethylated cytosine residues were then sulphonated by incubation in 3.12 mol/l sodium bisulphite (adjusted to pH 5.0) and 5 mmol/l hydroquinone (both Sigma) in a thermocycler (Hybaid; Thermo Life Sciences Ltd, Basingstoke, Hampshire, UK) for 15 seconds at 99°C and 15 minutes at 50°C for 20 cycles. Sulphonated DNA was then recovered using the Wizard DNA Cleanup system (Promega, Southampton, UK) according to the manufacturer’s instructions. The DNA was desulphonated by addition of 0.3 mol/l NaOH for 10 minutes at room temperature. The converted DNA was then ethanol precipitated and resuspended in water.
The BHD CpG island region was predicted using the UCSC genome browser (http://genome.ucsc.edu) and CpG plot (http://www.ebi.ac.uk). Both programs predicted a strong CpG island (observed/expected >0.60, %C+% G >60.00 and a length >200 bps) within the region −108bp to +854bp relative to the transcription start site of the BHD gene (AF517523). To analyse this CpG island region for the presence of methylation, specific PCR primers for the bisulphite modified sequence were designed.
The BHD CpG island spanning the first non-coding exon was analysed (fig 2) in colorectal and kidney tumour cell lines and kidney primary tumours using semi-nested PCR with the following primers; folliculin COBRA F (5′-TYGGYGGAATTATATTTAGAGTTTTTAAGTTT-3′) (forward) and folliculin COBRA R (5′-TACTTCCCAATCAAACCTCRAAATTATAA-3′) (reverse). One twentieth of this reaction was then used in a subsequent semi-nested PCR reaction using the primers folliculin COBRA F as before and folliculin COBRA Rnested (5′-TCRAACTATATTCCTAAACTTACRAAAACTAAA3′) (reverse) (where Y = C or T, and R = A or G, to ensure no bias towards methylated or unmethylated templates). The PCR conditions used in both reactions were: 95°C for 10 minutes followed by 25 cycles of denaturation for 1 minute at 94°C, annealing for 1 minute at 54°C and extension for 2 minutes at 74°C, and 35 cycles were used in the semi-nested PCR reaction. PCR products were then visualised on a 2% agarose gel with added ethidium bromide followed by incubation with the restriction endonucleases TaqI and BstUI for 2 hours at 65°C and 60°C respectively to assay for methylation. Two kidney and two colorectal cell line PCR products were randomly chosen for sequencing to confirm the specificity of the PCR reaction and to confirm the presence or absence of methylation. Prior to sequencing, PCR products were concentrated and purified using QIAquick PCR Purification columns (Qiagen, Crawley, West Sussex, UK) according to the manufacturer’s instructions. Purified PCR products were directly sequenced using the ABI BigDye cycle sequencing kit on an ABI 377 DNA sequencer (both Perkin Elmer). Sss1 in vitro methylated normal blood DNA was used as a positive control.
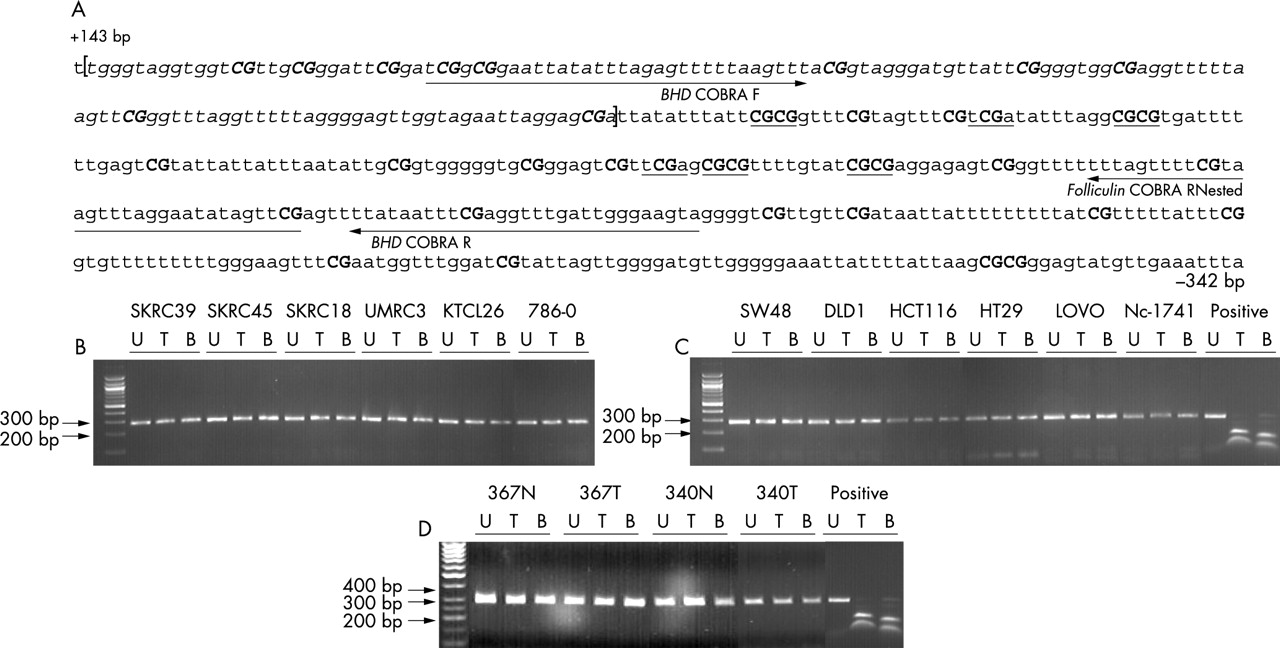
COBRA methylation analysis of the BHD CpG island. (A) The bisulphite modified sequence of the methylated BHD CpG island is shown. All non-CpG cytosines are converted to thymine, whereas cytosines within CpG dinucleotides remain as cytosine. Italic text represents exon 1 of the BHD gene. Underlined are the TaqI (tcga) and BstUI (cgcg) sites used to assay for methylation. Numbered are the first and last nucleotides of the sequence relative to the transcription start site of the BHD gene (GenBank no. AF517523). (B–D) Representative data of TaqI and BstUI restriction analysis of the BHD CpG island COBRA PCR product in kidney (B), colorectal tumour cell lines (C), and kidney tumours and respective normal tissue (D) following bisulphite treatment. The absence of digestion products shows that restriction sites have been lost due to bisulphite conversion and were therefore originally unmethylated. All cell lines analysed were unmethylated. Subsequent sequencing of two kidney and two colorectal tumour cell lines confirmed the COBRA results and the specificity of the PCR product. Sss1 in vitro methylated normal blood DNA was used as a positive control.
Statistical analysis
Fisher’s exact test and χ2 testing were used as appropriate. P values of <0.05 were taken as statistically significant.
RESULTS
BHD mutation analysis in renal cell carcinoma
SSCP analysis followed by sequencing of variant exons in 30 kidney cancer cell lines and tumours revealed two missense substitutions. An exon 12 somatic c.1785 G→T transversion producing a non-conservative missense Ala444Ser substitution was detected in a clear cell RCC tumour without a VHL mutation. Only the mutant sequence was present in the tumour (consistent with loss of the remaining wild type allele). In addition, a missense substitution was identified in one of the RCC cell lines: a conserved Ala238Val (c.1168 C→T) substitution in SKRC39, in which only mutant sequence was detected. In the absence of matching normal tissue DNA, it was not possible to define whether this change was somatic or germline; however, it was not detected in 80 control chromosomes.
BHD mutation analysis in colorectal cancer
SSCP analysis followed by sequencing of variant exons in 35 CRC cell lines and tumours revealed a germline missense substitution in 1/29 primary tumours and none of the cell lines. The c.1414 G→A transition resulted in a non-conservative Arg320Gln substitution, and both this and the wild type sequences were present in blood and tumour DNA. This change was not detected in 80 control chromosomes. The patient’s notes were reviewed. Age at diagnosis of the sigmoid CRC was 93 years and there was no evidence of microsatellite instability. There was no family history of CRC and no record of skin papules nor a history of RCC or pneumothorax (although these features had not been sought specifically).
In addition, a somatic c.1629 C→G transversion was demonstrated in a primary colorectal tumour. This produced a non-conservative missense substitution Arg392Gly, but was not associated with loss of heterozygosity and there was no evidence of microsatellite instability.
To determine if the germline Arg320Gln missense substitution might represent a low penetrance CRC susceptibility allele, exon 9 was sequenced in 159 cases of sporadic CRC (n = 75) and familial non-HNPCC CRC (n = 84). No Arg320Gln substitutions were identified, but an intron 9 splice site sequence variant (c.1517+6 G→A) was detected. Twenty-four of 159 (15%) CRC cases were homozygous for the A allele (16/75 (21%) sporadic cases and 8/84 (10%) familial cases), 57 (36%) were heterozygotes (23/75 (31%) sporadic cases, 34/84 (40%) familial cases) and 78 (49%) were GG homozygotes. The corresponding figures for 40 controls were 25% (n = 10) AA, 30% AG (n = 12) and 45% (n = 18) GG. While there was no difference between the allelic frequencies in normal controls and sporadic cases, there were fewer than expected AA homozygotes in the familial cases compared with controls (odds ratio 0.316; 95% CI 0.11 to 0.88; p = 0.03).
Two further exonic sequence changes were detected in the 29 colorectal tumour samples: a c.157 C→T transition was detected in the non-coding exon 1 in tumours and in normal controls, and a silent exon 7 c.1181 A→T transversion (Thr242Thr) was detected in the germline of two CRC patients.
Identification of intronic SNPs
In addition to the exonic changes described, a number of intronic SNPs were detected in patients and normals (table 1).
Non-coding sequence variants detected in a panel of kidney and colorectal cell lines and tumours and in normal controls.
BHD is not transcriptionally silenced in colorectal and renal tumour cell lines
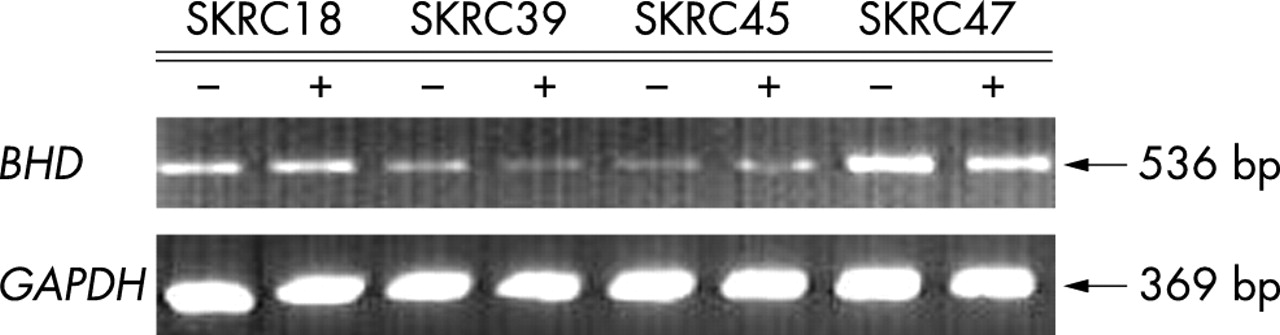
BHD promoter region methylation analysis was undertaken by COBRA assay and confirmed by sequencing. The CpG island analysed was located within the region −108bp to +854bp relative to the transcription start site (fig 2). We found no evidence of BHD CpG island methylation in 26 RCC, cell lines (n = 6) and primary tumours (n = 20), or in 12 CRC cell lines (fig 2). In addition we analysed BHD expression in six RCC and 12 colorectal cell lines after treatment with 5-aza-2′-dc. None of the cell lines demonstrated transcriptional silencing of BHD and there were no significant changes in expression (<10%) after treatment with 5-aza-2′-dc (fig 3).
{kind=link}
{kind=link}
{kind=link}
BHD expression by RT-PCR in kidney tumour cell lines (SKRC18, SKRC39, SKRC45 and SKRC 47), without (−) and with (+) 5-aza-2′-deoxycytidine (5-aza-dc) treatment. Cells were treated for 5 days with 5-aza-dc and expression was analysed by RT-PCR. All cell lines tested demonstrated no significant changes in BHD expression before and after 5-aza-dc treatment (BHD expression levels were normalised against GAPDH expression).
DISCUSSION
We found that somatic BHD inactivation occurred in a subset of sporadic clear cell RCCs. Thus, a somatic non-conservative substitution occurred in a sporadic clear cell RCC. This tumour also demonstrated loss of the wild type allele, consistent with a two hit mechanism of tumorigenesis. This mechanism has also been described in BHD associated RCC. The nature of the missense substitution in the SKRC 39 RCC cell line is unclear. It is not possible to determine whether this a somatic or germline event, but we note that there was no evidence of wild type sequence, therefore if the missense substitution is pathogenic, SKRC39 will not contain normal folliculin protein.
SKRC39 is a clear cell RCC cell line. Whereas specific histopathological subtypes of RCC are associated with germline VHL and MET gene mutations, and somatic VHL and RET mutations are seen in the corresponding sporadic tumours, a variety of RCC subtypes are reported in patients with BHD. Thus, Pavlovich et al11 found that 34% of BHD associated tumours were chromophobe RCC, 50% were hybrid oncocytoma/chromophobe RCC and 9% were clear cell (conventional) RCC. Although epigenetic silencing is now recognised as an important cause of tumour suppressor gene inactivation, and for genes such as RASSF1A is the predominant cause of inactivation, we did not find evidence of transcription silencing of BHD in clear cell RCC or of BHD promoter methylation in primary clear cell RCC. However, more extensive analysis of other RCC subtypes (for example, chromophobe RCC and oncocytoma) should be undertaken before concluding that epigenetic inactivation of BHD does not occur in RCC.
We identified a somatic missense substitution (Arg392Gly) in a primary CRC and a germline missense substitution (Arg320Gln) in another primary tumour. In both cases there was no evidence of loss of the wild type allele so it is not possible to conclude that there was complete inactivation of the folliculin protein in these tumours. Although the Arg320Gln variant was not detected in 80 control chromosomes, we cannot exclude that this is a polymorphism rather than a pathogenic mutation. The BHD gene contains a monunucleotide tract (C)8 in exon 11, which represents a mutational hot spot in BHD kindreds.15,16 However, we did not identify somatic BHD mutations related to the tract. Approximately 15% of sporadic CRCs have microsatellite instability (MSI) and mononucleotide tracts in such tumours frequently demonstrate alterations. All of the six CRC cell lines examined by us have been demonstrated to exhibit MSI, but recently Shin et al22 have reported BHD frameshift mutations in a subset (∼20%) of MSI+ CRCs.
The BHD gene product, folliculin, has no known function at present and so functional assays are not available to investigate the pathogenicity of the germline Arg320Gln missense substitution identified in this study. However, the identification of further BHD missense mutations in BHD kindreds and sporadic tumours may provide insights into folliculin function by identifying critical domains for normal tumour suppressor activity.
Common sequence variants in tumour suppressor genes provide good candidates for low penetrance susceptibility alleles. It is intriguing that we found an intron 9 sequence variant to be more frequent in CRC cases than in controls (p = 0.055). The G→A transition is not predicted to significantly influence splicing efficiency (http://www.cbs.dtu.dk/services/NetGene2/), so it may be that the intron 9+6 G→A variant is in linkage dysequilibrium with other pathogenic variants. Nevertheless, our results suggest that further investigation of the role of BHD sequence variants in CRC susceptibility may be indicated.
Acknowledgments
We thank Cancer Research UK for financial support.