Article Text

Abstract
Background Exonic variants of unknown biological significance (VUS) identified in patients can affect mRNA splicing, either by changing 5′ or 3′ splice sites or by modifying splicing regulatory elements. Bioinformatic predictions of these elements are still inaccurate and only few such elements have been functionally mapped in BRCA2. We studied the effect on splicing of eight exon 7 VUS, selected from the French UMD-BRCA2 mutation database.
Methods We performed splicing minigene assays and analyses of patient RNA. We also developed a pyrosequencing-based quantitative assay, to measure, in patient RNA, the relative contribution of each allele to the production of exon 7-containing transcripts. Moreover, an exonic splicing enhancer (ESE)-dependent minigene assay was used to evaluate the splicing regulatory properties of wild-type and mutant segments.
Results Six out of the eight variants induced splicing defects. In the minigene assay, c.517G>T and c.631G>A altered the natural splice sites, c.572A>G created a new 5′ splice site, and c.520C>T, c.587G>A and c.617C>G induced exon 7 skipping (66%, 25% and 46%, respectively). Pyrosequencing of patient RNA confirmed these levels of exon skipping for c.520C>T and c.617C>G. Results from the ESE-dependent minigene assay indicated that c.520C>T and c.587G>A disturb splicing regulatory elements.
Conclusions BRCA2 exon 7 splicing is regulated by multiple exonic elements and is sensitive to disease-associated sequence variations. Measurements of allelic imbalance in patient-derived RNA and/or quantitative analyses using minigene assays provide valuable estimates of the extent of partial splicing defects. Assessment of pathogenicity of variants with partial splicing effect awaits additional evidence and especially the completion of segregation analyses.
Statistics from Altmetric.com
Introduction
The interpretation of the numerous sequence variants of unknown biological and clinical significance (VUS) identified in patients during genetic screenings represents a major challenge in medical genetics. Clinical management and genetic counselling is often limited by lack of knowledge about the pathogenic character of these VUS. This problem is particularly important in the molecular diagnosis of hereditary breast and ovarian cancers, given that the cancer susceptibility genes, BRCA1 and BRCA2, exhibit a large spectrum of sequence variations.1 The French UMD-BRCA2 mutation database includes a total of 1735 distinct variants, of which 1089 are VUS (http://www.umd.be/BRCA2/).2
In principle, VUS can be classified using an integrated evaluation approach based on multifactorial likelihood prediction models, which applies a Bayesian statistical inference to each VUS.3–5 This approach takes into account patient and family history and cosegregation data combined with the predicted effect of the VUS at the protein level. However, it is presently limited by not being able to fully integrate the potential effect of the VUS at the mRNA level.6–8
It is now well established that exonic variations can disrupt pre-mRNA splicing.9–12 Sequence variations may affect splicing by altering 5′ or 3′ splice site canonical sequences (5′ ss or 3′ ss) through destruction of the natural sites and/or creation of a new site and/or activation of a cryptic site. In addition, exonic variations may alter splicing by modifying splicing regulatory elements, such as exonic splicing enhancers and/or silencers (ESE and ESS), or by affecting RNA secondary structure.13
Although several bioinformatic tools are currently available to predict the effect of a variant on splicing, these tools are not fully accurate and need improvement, in particular those assessing splicing regulatory elements. Considerable progress in this field is expected to arise from high-throughput mutational analyses,14 as well as from bio-statistical studies aiming to determine the genome-wide positional distribution of regulatory elements.15 Large sets of experimental data derived from the analyses of disease-associated VUS will be crucial to transform these studies into validated splicing prediction programmes.
We have previously shown that the BRCA2 c.520C>T variant induces exon 7 skipping.16 Here, we investigated the effects on splicing of seven additional VUS located in this exon and identified four new splicing mutations. This study provides evidence that the inclusion of BRCA2 exon 7 requires a positive regulation, involving multiple ESEs and that this exon is particularly sensitive to splicing mutations.
Methods
Patient genetic analysis
The BRCA2 VUS tested in this study (table 1) were detected in probands selected from families undergoing genetic counselling in the BRCA diagnostic laboratories from the French Unicancer Genetic Group (UGG). The criteria for diagnostic mutation screening of the BRCA2 gene in patients were applied according to the current French recommendations. Screening for BRCA2 mutations was performed as previously described.16 Informed consent was obtained from all patients.
Nomenclature
The DNA sequence numbering is based on the cDNA sequence of BRCA2 (NCBI RefSeq NM_000059.3), following the recommendations of the Human Genome Variation Society (first position of the translation initiation codon ATG denoted as c.1).
Bioinformatic predictions of splicing alterations
Bioinformatic prediction tools were used to evaluate the effect of selected VUS on the canonical 3′ and 5′ splice sites and on splicing regulatory elements, as described under supplementary materials.
Cell culture
HeLa cells (human cervical adenocarcinoma epithelial cell line) were purchased from ATCC. HBL-100 (immortalised human normal breast epithelial cell line)17 and MCF-7 (human breast adenocarcinoma epithelial cell line)18 were provided by S. Mazoyer (CRCL, Lyon, France) and L. Poulain (BioTICLA, Caen, France), respectively. All cell lines were grown in Dulbecco's modified Eagle medium (Gibco) supplemented with 10% fetal calf serum in a 5% CO2 atmosphere at 37°C.
Splicing minigene reporter assay
The splicing minigene assay has been previously described,19–22 and was performed as follows.
Generation of minigene constructs
Wild-type and mutant genomic segments were amplified by PCR from patient genomic DNA using forward primer BR2-7-Bgl-F (5′-GACCAGATCTGTTATACCTTTGCCCTGAGATTTA-3′) and reverse primer BR2-7-Mlu-R (5′-GACCACGCGTGCTTGACACCACTGGACTACC-3′), carrying 5′ tails with BglII and MluI restriction sites, respectively (underlined). The PCR-amplified genomic segments encompassed the exon 7 (115 bp) of BRCA2 and part of the flanking intronic 5′ and 3′ regions (159 and 207 bp, respectively). After digestion with BglII and MluI, the PCR products were inserted into the BamHI and MluI cloning sites in the intron of pCAS2, a two-exon splicing reporter minigene vector (figure 1B). The insert was then sequenced to identify the wild-type and variant minigene constructs and to ensure that no extra mutations were added during amplification or cloning.

Analysis of eight selected BRCA2 exon 7 variants of unknown biological and clinical significance (VUS) by using a functional splicing minigene reporter assay. (A) Positions of the selected BRCA2 exon 7 VUS. The nucleotide and amino-acid sequences of BRCA2 exon 7, including the three flanking intronic nucleotides are shown. The boxes above the sequence indicate the position of each VUS. Numbers above and below the sequence refer to cDNA sequence and to codon numbering, respectively. (B) Schematic representation of the pCAS2-BRCA2-exon7 minigenes. Boxes represent the exons and the lines in between symbolise the introns. Genomic fragments containing the exon 7 of BRCA2 and flanking intronic sequences, either wild-type or mutant, were inserted into the pCAS2 minigene as indicated and described under Methods. Transcription is driven by the human cytomegalovirus immediate-early promoter (CMV) as indicated. Primers used for RT-PCR analysis are represented by arrows. (C) Effect on exon 7 splicing of the selected VUS assessed by using the functional splicing minigene assay. Minigene pCAS2 constructs, carrying no insert (vector, V) or either carrying the wild-type (Wt) or the mutant BRCA2 exon 7, were transiently expressed in HeLa cells. The splicing patterns of the minigenes were monitored by RT-PCR as described under Methods. An image of an ethidium bromide-stained gel is shown. The positions of the RT-PCR products with (+) or without (Δ) BRCA2 exon 7 are indicated on the right. At least three independent transfections were performed for each minigene construct. a, Transcripts with exon 7 deleted of the last 60 nucleotides; b, Transcripts with exon 7 deleted of the last 70 nucleotides.
Transfection and RT-PCR analysis
The wild-type and mutant minigene constructs were transiently transfected into HeLa cells using the FuGENE 6 transfection reagent, according to manufacturer's instructions (Roche Applied Science). Cells were then collected 24 h post-transfection. Total RNA was extracted using the NucleoSpin RNA II kit (Macherey Nagel), according to the manufacturer's instructions, including a DNase treatment. The RT-PCR reactions were performed using the OneStep RT-PCR kit (Qiagen), according to the manufacturer's instructions, with 200 ng RNA as template in a 25 µl reaction volume. Reactions were performed using the forward primer KO1F (5′-TGACGTCGCCGCCCATCAC-3′) and the reverse primer pCAS2R (5′-ATTGGTTGTTGAGTTGGTTGTC-3′) (figure 1B), with 30 cycles of amplification. RT-PCR products were separated by electrophoresis on 2% agarose gels containing ethidium bromide and visualised by exposure to ultraviolet light under conditions of non-saturating exposure. Semi-quantitative analysis, gel extraction and sequencing of the RT-PCR products were carried out as previously described.22
ESE-dependent splicing assay
Generation of pcDNA-Dup constructs
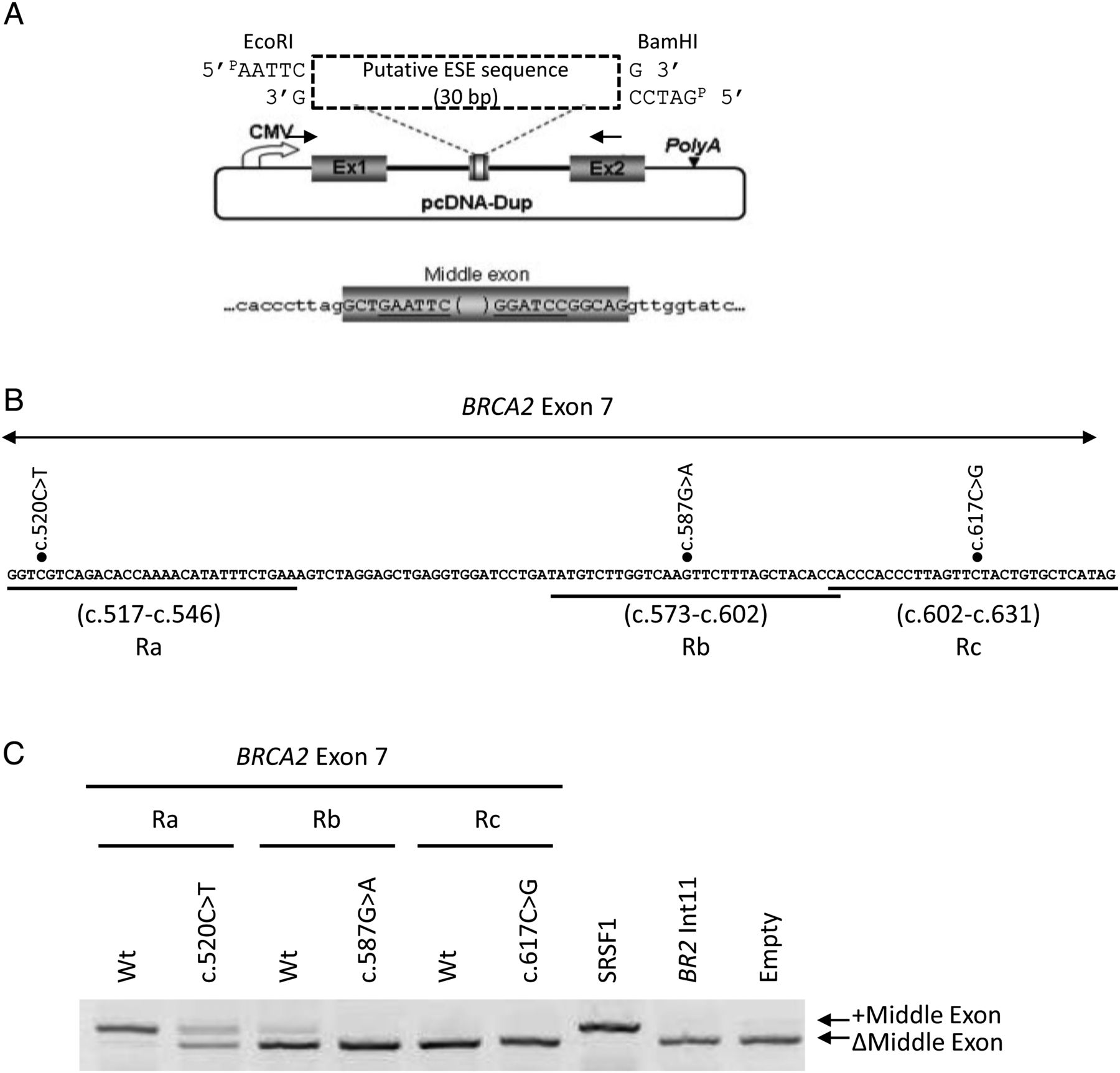
Three 30-bp long segments of BRCA2 exon 7, corresponding to regions encompassing nucleotides c.517-c.546 (Region a, Ra), c.573-c.602 (Rb) or c.602-c.631 (Rc) (figure 3B), were tested for the presence of splicing enhancer (ESE) properties by using an ESE-dependent splicing minigene assay, as previously described (figure 3A).19 ,22 Briefly, the exonic fragments of interest were obtained by annealing complementary 5′-phosphorylated oligonucleotides carrying 5′-EcoRI and 3′-BamHI compatibles ends. Duplexes were inserted into the EcoRI and BamHI cloning sites in the middle exon of the pcDNA-Dup plasmid. This vector contains a splicing cassette consisting of a β-globin-derived three-exon minigene (Dup) under the control of the cytomegalovirus (CMV) promoter (figure 3A). The effects of the mutations BRCA2 c.520C>T, c.587G>A and c.617C>G were tested in parallel in the relevant segments. Control experiments were performed, as previously described.19 ,22
Transient transfection and RT-PCR analysis
Different pcDNA-Dup constructs were transiently transfected into HeLa cells and total RNA was extracted 24 h post-transfection, as described above for the splicing minigene reporter assay. RT-PCR reactions were performed using the OneStep RT-PCR kit (Qiagen), according to the manufacturer's instructions, with 150 ng RNA as template in a 25 µl reaction volume. RT-PCR reactions were performed using previously described primers T7-Pro and Dup-2R,19 with 28 cycles of amplification. RT-PCR products were separated by electrophoresis on a 2.5% agarose gel containing ethidium bromide and visualised by exposure to ultraviolet light. Gel extraction and sequencing of the RT-PCR products were carried out as previously described.22
RT-PCR analysis of patient RNA
Whole blood samples were collected from patients and voluntary healthy donors in PAXgene Blood RNA tubes (Qiagen). RNA extraction was performed according to the manufacturer's protocol, including DNase treatment. RT-PCR was performed from 250 ng of total RNA using the One-step RT-PCR kit (Qiagen). Two sets of primers were used: one set with the forward primer BR2-6-RT-F (5′-TGGTATGTGGGAGTTTGTTTC-3′), complementary to BRCA2 exon 6, and the reverse primer BR2-9-RT-R (5′- CATGACTTGCAGCTTCTCTTT-3′), complementary to BRCA2 exon 9, and another set with the forward primer BR2-3-RT-F (5′-CTGCCGCTGTACCAATCTC-3′), complementary to BRCA2 exon 3, and the reverse primer BR2-8-RT-R (5′-TTCAGATGCTTCTTCATTT-3′), complementary to BRCA2 exon 8. All amplifications were performed using a touch-down PCR programme as previously described.22 RT-PCR products were separated by electrophoresis on 0.8% agarose gels containing ethidium bromide and visualised by exposure to ultraviolet light. RT-PCR products were gel-purified and sequenced as previously described.22
Allele-specific expression analysis in patient peripheral blood
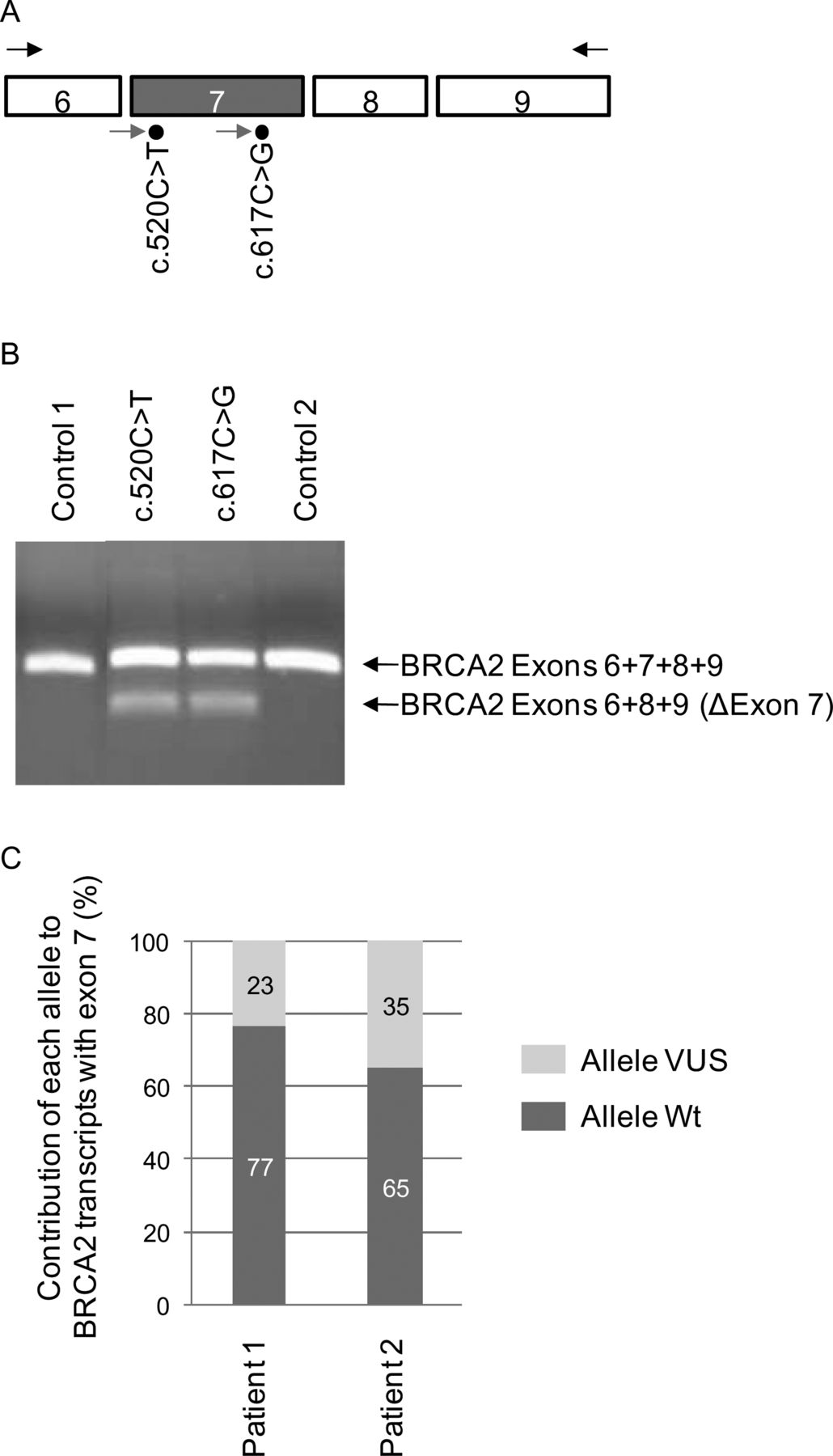
Allele-specific expression of BRCA2 was evaluated by pyrosequencing, using a PyroMark Q24 system (Qiagen, France). RT-PCR products were obtained with the same protocol as described above under RT-PCR analysis of patient RNA, except that RT-PCR was performed from 100 ng of total RNA, using a 5′ biotinylated reverse primer and the PCR programme had 40 cycles of amplification: 94°C, 30 s; 50°C, 30 s; 72°C, 1 min. Fifteen µl of the PCR product were sequenced according to the protocol of the PyroMark Q24 system manufacturer with 1.2 pmol of specific pyrosequencing primers: BR2pyro520F (5′-CCAAAGTTTGTGAAGGG-3′) or BR2pyro617F (5′-CACCACCCACCCTTA-3′) for the variants c.520C>T and c.617C>G, respectively (figure 2A). Pyrograms were analysed with the software PyroMark Q24 V.2.0.6 (Qiagen, France), in order to determine the relative contribution of both alleles, wild-type and mutant, to the production of BRCA2 transcripts containing exon 7.

The BRCA2 c.520C>T and c.617C>G variants are associated with abnormal splicing in patient RNA. (A) Positions of primers used in RT-PCR and pyrosequencing experiments. Exons 6 to 9 of BRCA2 are represented as boxes. Primers used for the RT-PCR analyses of patient blood cell RNA are represented by the arrows above exons 6 and 9. Primers used for pyrosequencing are indicated by arrows below exon 7 (grey box). (B) BRCA2 exon 7 splicing alteration detected in patient peripheral blood. The splicing patterns were monitored by RT-PCR using primers described above and RNA from patients carrying either the BRCA2 c.520C>T or c.617C>G variants and from two control individuals. The image of the ethidium bromide-stained gel is representative of two independent experiments. The identity of the transcripts is indicated on the right. (C) Relative contribution, in patient peripheral blood, of the wild-type (Wt) and mutant BRCA2 alleles to the production of exon 7-containing transcripts, as determined by pyrosequencing. Patients 1 and 2 carry BRCA2 variants c.520C>T and c.617C>G, respectively. RT-PCR and pyrosequencing were performed in two independent experiments, each experiment made in triplicate. SDs were SD=±2.0 and SD=±2.5 for patients 1 and 2, respectively.
Results
Sequence variants in BRCA2 exon 7 selected from the French UMD database
This study was prompted by our previous finding that the BRCA2 VUS c.520C>T induced strong exon 7 skipping in a minigene assay.16 Here, we selected seven additional VUS located in exon 7, from the French UMD-BRCA2 mutation database (http://www.umd.be/BRCA2/)2 (table 1, figure 1A). This selection included the six missense variants, c.517G>T (p.Gly173Cys), c.572A>G (p.Asp191Gly), c.587G>A (p.Ser196Asn), c.617C>G (p.Ser206Cys), c.625C>T (p.Leu209Phe), c.631G>A (p.Val211Ile), and the synonymous variant, c.522T>G (p.Arg174Arg). All these variants were identified in patients undergoing molecular diagnosis in the BRCA laboratories from the French UGG. The patients carrying these variants were referred for genetic counselling based on patient and family history (table 1).
Family data for the BRCA2 exon 7 variants analysed in this study
Bioinformatic tools predict splice site alterations for three out of the eight selected BRCA2 exon 7 VUS
We evaluated the effect of each VUS on the strength of the natural splice sites, as well as on the potential creation of alternative splice sites, by using four bioinformatic programmes (SSF, MaxEntScan, NNSPLICE and HSF) (see supplementary materials). This analysis allowed us to identify three variants for which an alteration in splice site definition was predicted: c.517G>T, c.572A>G and c.631G>A (see supplementary table S1 and figure S1). The first variant, c.517G>T, located at position +1 of the exon, was predicted by all algorithms to slightly decrease the strength of the natural 3′ ss. The second variant c.572A>G, located in the centre of exon 7 at position +56, was not predicted to modify the canonical splice sites, but, according to the four algorithms, it creates a 5′ ss at position c.571, with a higher strength than the one estimated for the physiological 5′ ss. The third variant c.631G>A, located at the last nucleotide of exon 7, was predicted to either abolish (MaxEntScan) or diminish the strength of the 5′ ss (SSF and HSF). No alteration of splice sites were predicted for the other five selected VUS, c.520C>T, c.522T>G, c.587G>A, c.617C>G and c.625C>T (see supplementary table S1).
We also analysed each VUS for its potential effect on splicing regulatory elements using three additional bioinformatic tools (see supplementary materials, supplementary tables S2 and S3). Overall, the results obtained with these tools were inconclusive because highly variable and difficult to interpret (see supplementary tables S2 and S3).
Identification of new splicing mutations in BRCA2 exon 7 by using a splicing minigene assay
We performed functional splicing minigene assays to experimentally determine the effects on splicing produced by the eight BRCA2 exon 7 VUS. Our results show that six out of the eight variants induce splicing defects (figure 1C). As predicted by in silico analysis (see supplementary table S1), c.517G>T, c.572A>G and c.631G>A had a direct effect on the definition of splices sites. Variant c.517G>T induced total exon skipping (RNA r.517_631del), a defect that results in a frame shift (p.Gly173SerfsX19) (figure 1C and supplementary figure S1). Variant c.572A>G produced mainly a transcript containing only a portion of exon 7, deleted for the last 60 nucleotides (RNA r.572_631del). This result is in good agreement with the predicted creation of a 5′ ss at position c.571 (figure 1C, supplementary table S1, figure S1). This splicing defect creates an in-frame deletion (p.Asp191_Ile210del). Variant c.631G>A, at the last nucleotide of exon 7, mainly induced exon skipping, which is concordant with the predicted destruction or decrease of the 5′ ss strength (figure 1C, supplementary table S1, figure S1). This variant also produced a minor transcript, corresponding to the inclusion of exon 7 deleted of the last 70 nucleotides (RNA r.562_631del), as a consequence of the activation of a cryptic 5′ ss at position c.561 (figure1C, supplementary table S1, figure S1). This deletion is predicted to result into a frame shift (p.Val188SerfsX19).
In the same assay, c.522T>G and c.625C>T displayed reproducibly the same splicing pattern as the wild-type, indicating that these variants do not have an effect on splicing in the minigene context. In contrast and as previously reported,16 c.520C>T induced the exclusion of exon 7, here evaluated at 66% (figure 1C). As shown on figure 1C, two additional VUS, c.587G>A and c.617C>G induced exon 7 skipping, but to a different extent, 25% and 46%, respectively (figure 1C).
In order to investigate the effect of the cellular context on the level of exon skipping of these three variants, we performed the minigene assay in two additional cell lines: the immortalised human normal breast epithelial cell line HBL-100 and the human breast adenocarcinoma cell line MCF-7. Our results revealed that, in these cell lines, the c.520C>T, c.587G>A and c.617C>G variants appeared to induce levels of exon 7 skipping similar to those observed in HeLa cells (see supplementary figure S2).
The BRCA2 c.520C>T and c.617C>G variants are associated with abnormal splicing in patient RNA
We then decided to verify if the splicing defects observed in the minigene assay could also be detected in a physiological context. As shown on figure 2, we analysed by RT-PCR the splicing patterns of BRCA2 from patients carrying variants c.520C>T and c.617C>G. Blood samples compatible with RNA extraction were not available from patients carrying the other selected variants. Results were compared to those obtained with two control individuals (figure 2B). A single product with the expected size (318 bp) was detected in the control subjects. Sequencing analysis confirmed that it corresponded to the region covering exons 6 to 9 of the BRCA2 transcript (figure 2B). In contrast, the analysis of the RNA from patients carrying either c.520C>T or c.617C>G revealed the presence of an additional RT-PCR product with a smaller size (203 bp) that was identified by sequencing as an aberrant BRCA2 transcript lacking exon 7 (figure 2B). These results confirm that these variants induce exon 7 skipping in a physiological context. For both patients, the levels of aberrant transcripts without exon 7 were lower than those of transcripts containing exon 7. Sanger sequencing of the RT-PCR products containing exon 7 revealed the presence of both wild-type and mutant sequences (data not shown). Then, in order to quantitatively evaluate the contribution of wild-type and mutant alleles to the production of BRCA2 transcripts containing exon 7, we took advantage of the quantitative nature of pyrosequencing and determined the relative amount of each allele within the RT-PCR product containing exon 7. This approach allowed us to determine that, in patient peripheral blood cells, the BRCA2 c.520C>T and c.617C>G mutant alleles contributed only to 23% and to 35%, respectively, of the total levels of BRCA2 transcripts containing exon 7 (figure 2C). Assuming that both alleles, wild-type and mutant, are transcribed at the same level, these data are in good agreement with results obtained by using the monoallelic splicing minigene reporter assay (figure 1C). More precisely, in these conditions, one can deduce that the relative amount of transcripts with and without exon 7 is, respectively, 30% and 70% for c.520C>T and 54% and 46% for c.617C>G.
Because an alternative splicing event corresponding to the skipping of both exon 6 and exon 7 is often observed in BRCA2 transcripts (our unpublished data), we also performed RT-PCR analysis using a forward primer located in exon 3 and a reverse primer located in exon 8. In addition to the main RT-PCR product corresponding to the region covering exons 3 to 8, a BRCA2 isoform lacking both exon 6 and exon 7 was detected in control subjects, as well as in the patients carrying the variants c.520C>T or c.617C>G (see supplementary figure S3). However, a third band corresponding to skipping of exon 7 alone was detected in the two patient samples but not in the control subjects. These results indicate that these variants specifically induce exon 7 skipping, without a noticeable effect on the alternative splicing event involving both exons 6 and 7 (see supplementary figure S3).
Variants c.520C>T and c.587G>A alter enhancer properties of two distinct regions of BRCA2 exon 7
The variations c.520C>T, c.587G>A and c.617C>G are located, respectively, in the 5′ region (position +4 of the exon), in the central region (position +71) and in the 3′ region (position -15 relative to the end of the exon) of BRCA2 exon 7 (115 bp) (figure 3B). These VUS are therefore positioned outside the sequences at intron-exon junctions that define the 3′ and 5′ splice sites.9 It is possible that these mutations induce exon skipping by affecting splicing regulatory elements, either by disrupting an ESE and/or by creating an ESS. In order to map potential regulatory elements, we took advantage of the previously described ESE-dependent splicing assay.19 ,22 This assay is based on the analysis of the splicing behaviour of the middle exon of pcDNA-dup (figure 3A), a three exon minigene that is particularly sensitive to splicing regulatory elements. Because of a weak 3′ ss, the middle exon of pcDNA-dup is not recognised by the splicing machinery unless enhancer elements are inserted within its sequence. Representative minigenes were constructed by inserting 30 bp-long DNA fragments encompassing each mutation (segments Ra, Rb and Rc), into the middle exon of pcDNA-dup (figure 3B). As shown on figure 3C, the wild-type Ra segment, corresponding to the 5′ region of BRCA2 exon 7 (c.517-c.546), was able to induce strong inclusion of the middle exon, suggesting that this region contains a sequence with enhancer properties. Introduction of the mutation c.520C>T into this fragment induced a major exclusion of the middle exon (figure 3C). We also found that the wild-type Rb fragment (c.573-c.602) had a weak but reproducible positive effect on middle exon inclusion and that the c.587G>A mutation abolished this effect. These data suggest that c.520C>T and c.587G>A modify distinct exonic splicing regulatory elements. In the same assay, the wild-type and mutant (c.617C>G) Rc segments, corresponding to the 3′ region of exon 7 (c.602-c.631), did not induce the inclusion of the middle exon (figure 3C), indicating the absence of ESE elements in this region and suggesting that c.617C>G may induce exon 7 skipping by creating an ESS.
{kind=link}
{kind=link}
{kind=link}
Variants c.520C>T and c.587G>A alter enhancer properties of two distinct regions of BRCA2 exon 7. (A) Schematic representation of the pcDNA-Dup minigene used in the ESE-dependent splicing assay. The plasmid pcDNA-Dup has a pcDNA3.1(−) backbone and a β-globin-derived three exon minigene (grey boxes) under the control of the cytomegalovirus (CMV) promoter. The middle exon is flanked upstream and downstream by the 130 nucleotides of human β-globin intron 1. The sequence of the intron-exon boundaries of the middle exon are shown, with intronic and exonic sequences indicated in lower and upper case, respectively. Different 30 bp segments of BRCA2 exon 7 were inserted into the EcoRI and BamHI sites of the middle exon, as indicated. Primers used for RT-PCR analysis are represented by arrows. (B) BRCA2 exon 7 segments tested in the ESE-dependent splicing minigene assay. The 30 bp-long segments analysed correspond to the exonic regions (R) that encompass each of the three mutations of interest. The nucleotide sequence of BRCA2 exon 7 is shown. The position of the exonic segments of interest, (c.517-c.546) (Ra), (c.573-c.602) (Rb) and (c.602-c.631) (Rc), is indicated underneath the sequence. Black dots indicate the variants tested in this assay. (C) ESE-dependent splicing minigene assay. The 3 exonic fragments (Ra, Rb, Rc), either wild-type or mutant as indicated, were tested in parallel with control pcDNA-dup minigenes, as previously described.19 The pcDNA-dup positive control contained a triplet of binding sites for the splicing regulatory protein SRSF1. One negative control contained an arbitrary fragment without known regulatory elements derived from BRCA2 intron 11 (BR2 Int11), whereas the other did not contain any insert (Empty). The minigenes were transfected into HeLa cells and the transcripts were analysed by RT-PCR as described under Methods. A picture of an ethidium bromide-stained gel is shown. The positions of the RT-PCR products with (+) or without (Δ) the middle exon are indicated on the right. Results are representative of at least three independent experiments.
Discussion
In this study, we analysed the effect on splicing of eight VUS located in exon 7 of BRCA2. Our results show that six out of these variants induce splicing defects and that they can be separated into two distinct groups: (i) mutations that alter the intrinsic definition of splice sites, and (ii) mutations that alter splicing regulatory elements located at a distance from the splice sites.
The first group includes the variants c.517G>T, c.572A>G and c.631G>A. Variants c.517G>T and c.631G>A, located respectively at the first and last base of exon 7, lead to exon skipping because they modify the sequence of the 3′ and the 5′ splice sites, respectively. The drastic effect of c.517G>T is surprising given that the decrease in 3′ ss strength predicted by in silico tools is less than 10%, a value that is lower than the one often used as a threshold (20%) for stratifying VUS for splicing functional analysis.25 Variant c.631G>A also induces major exon skipping, a result that is in good agreement both with bioinformatic predictions and with data obtained from the analysis of patient RNA.23 ,24 Variant c.572A>G, located in the centre of exon 7, leads to the production of an aberrant transcript lacking 60 nt of the 3′ terminal portion of the exon, a result that is in good agreement both with the predicted creation of a 5′ ss at position c.571 and with data obtained from the analysis of patient RNA (D. Muller, Strasbourg, personal communication). It is presently unknown if this in-frame deleted mRNA is relevant to cancer predisposition. Overall, the data obtained from the analysis of these VUS: (i) confirm the value of bioinformatic predictions of splice site changes and (ii) raise the critical issue of score variation thresholds.
The second group of splicing mutations include variants c.520C>T, c.587G>A and c.617C>G. Our results, derived from minigene assays, indicate that these VUS induce different levels of exon 7 exclusion by modifying splicing regulatory elements. The splicing defects induced by these mutations were difficult to predict by using in silico tools, which provided, for the most part, results inconsistent with the data obtained experimentally. Our findings underscore the importance of performing functional assays to identify mutations altering splicing regulatory elements. Importantly, the effects of c.520C>T and c.617C>G were confirmed on patient RNA. Interestingly, two other mutations inducing partial exon 7 skipping have been recently described, c.518G>T (p.Gly173Val)26 and c.581G>A (p.Trp194X)27. Thus, our results bring up the number of mutations affecting exon 7 splicing regulation to a total of 5. This observation is remarkable taking into account the small number of variants identified thus far that affect the splicing regulation of BRCA2. To our knowledge, in the BRCA2 gene, besides the exon 7 mutations described above, only one mutation in exon 3, c.231T>G (p.Thr77Thr),16 and two mutations in exon 18, c.8165C>G (p.Thr2722Arg)28 and c.7992T>A (p.Ile2664Ile),16 have been reported to induce exon skipping by altering splicing regulatory elements. Our study indicates that the number of such mutations may be currently underestimated and points to BRCA2 exon 7 as the exon with the highest number of splicing regulatory mutations identified, thus far, in the BRCA genes.
The relative high number of disease-associated variants affecting the regulation of exon 7 splicing and the observation that these mutations are differently distributed within the exon suggest that this exon is regulated by multiple regulatory elements. The presence of multiple and sometimes overlapping splicing regulatory elements has been documented in several studies, for example, in exon 12 of the CFTR gene.29 Here, we identified two regions of BRCA2 exon 7 with potential ESE properties that are altered by disease-associated variants. It is possible that these variants destroy an ESE and/or create an ESS. Further studies will be required to clarify the mechanisms underlying the splicing defects produced by these variants.
Recent genome-wide analyses of hexameric motifs have shown that a large fraction of disease-causing exonic variants alters potentially functional splicing regulatory elements.14 ,15 This fraction has been estimated to be in the range of 20–25% of all reported missense and nonsense mutations.15 ,30 However, it is still impossible to predict in silico if the splicing of a particular exon depends on the presence of functional exonic regulatory elements or if a particular nucleotide change induces a splicing defect by altering this type of regulation. By performing an in silico analysis, we observed that the 5′ splice site score of exon 7 is below the average score of the entire set of BRCA2 5′ splice sites (data not shown). It has been previously suggested that exons with weak splice sites might require a stronger positive splicing regulation to ensure exon inclusion.31 This may be the case for BRCA2 exon 7. Based on this assumption, we propose that a special attention should be given to disease-associated VUS located in exons with weak splice sites.
Mutations with partial effects on splicing are often observed but these effects are usually not precisely quantified. It is currently argued that such precise quantitative measurements are needed in order to define, in the future, the appropriate prior probability scores for splicing mutations;6 prior probabilities being an essential component of the multifactorial Bayesian approach to the assessment of pathogenicity.32 ,33 Here, we show that it is possible to perform an accurate quantitative evaluation of these defects by combining results from a minigene assay and measurements of allelic ratios in patient RNA using a pyrosequencing approach. On one hand, the minigene assay has the advantage of establishing a direct causality relation between a splicing defect and the presence of a variant. This assay allows the accurate measurement of the fraction of exon inclusion both because: (i) it is a monoallelic system and (ii) the minigenes used in our assay produce transcripts refractory to nonsense mediated decay. On the other hand, pyrosequencing of patient RNA circumvents the problems of biallelic expression and of eventual instability of aberrant transcripts expressed from the mutant allele by focusing on the contribution of each allele to the production of exon 7-containing transcripts. The good correlation between these two approaches, used here to analyse variants c.520C>T and c.617C>G, suggests that both methods can be used to evaluate quantitatively the effect of any exonic sequence variation that results in partial exon skipping.
This study highlights the difficulty of defining the pathogenic character of mutations with a partial effect on splicing. The difficulty is due to several reasons. First, it remains to be known if the effect of such mutations, particularly those altering splicing regulation, is dependent on the cellular context. It would be of great interest to analyze the consequences of these mutations in normal and tumoral breast tissues of carrier individuals. However, these samples are usually not available. Our alternative preliminary approach to address this question was to perform minigene assays in two cell lines derived from breast epithelial tissues, either normal or tumoral, and to compare the results with those obtained with HeLa cells. We observed that the partial splicing defects induced by c.520C>T, c.617C>G and c.587G>A were similar in the three cell lines suggesting that the extent of exon 7 skipping caused by these variants is not attenuated in breast epithelial cells. Second, the critical level of functional BRCA2 protein needed to ensure efficient double-strand break repair and protection from tumour development is not known. It is possible that a decrease in the expression of BRCA2 becomes deleterious below a certain threshold. A dose-dependent effect has been described for instances for PTEN, another tumour suppressor gene, for which even subtle reductions of functional levels are sufficient to drive tumour initiation and progression.34 Third, a missense mutation with a partial effect on splicing can be deleterious because it potentially combines a RNA defect with a protein defect resulting from the missense alteration (encoded by the residual full-length transcripts). Three different algorithms were used to predict the effect of the selected VUS on protein function (see supplementary materials, supplementary table S4).
Finally, this study exemplifies how screening disease-associated VUS for splicing mutations allows the detection of previously unforeseen splicing regulatory elements. In the future, such approaches may contribute to improve the current in silico predictions of splicing regulatory elements. Moreover, this study contributes to the assessment of pathogenicity of BRCA2 VUS. We propose that the data obtained here, combined with results from upcoming co-segregation analyses, should be integrated into a Bayesian probability calculation in order to achieve a definitive classification of the BRCA2 variants described in this study.
Acknowledgments
We thank the patients and their families who have contributed to this project. We are grateful to the oncogeneticists from the French Unicancer Genetic Group, who provided patient genomic DNA and family data: Pascaline Berthet, Centre François Baclesse, Caen; Violaine Bourdon, Institut Paoli Calmettes, Marseille; Catherine Dugast, Centre Eugene Marquis, Rennes; Joelle Fournier, Centre Oscar Lambret, Lille; Marine Guillaud-Bataille, Institut Gustave Roussy, Villejuif; Mélanie Leone, Sylvie Mazoyer, Centre de Recherche en Cancérologie, Lyon; Virginie Moncoutier, Service d'Oncogénétique, Institut Curie, Paris; Danièle Muller, Centre Paul Strauss, Strasbourg; Audrey Remenieras, Institut Paoli-Calmettes, Marseille; Olga Sinilnikova, Centre de Recherche en Cancérologie, Lyon; Nancy Uhrhammer, Centre Jean Perrin, Clermont-Ferrand. We thank S. Mazoyer (CRCL, Lyon, France) and L. Poulain (BioTICLA, Caen, France) for generously providing the HBL-100 and MCF-7 cell lines, respectively. We thank A. Blavier and F. Wolinski (Interactive Biosoftware, Rouen, France) for their help in the use of the in silico prediction tools.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors PG, MT, AM and SK designed the study and wrote the manuscript; PG, SK, DDG, JA, FR, EJ, EB, DV, DL carried out the experiments; SC, ER, RL are the curators of the French BRCA database and coordinated sample collection. All authors read and approved the final version of the manuscript.
-
Funding This work was supported by a joint translational research grant of the French National Cancer Institute and the ‘Direction Générale de l'Offre des Soins’ (INCa-DGOS). P. Gaildrat was partially supported by the French North West Canceropole. P. Gaildrat, S. Caputo and J. Abdat were supported by the French National Cancer Institute (INCa).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.