Article Text

Abstract
Background: The chromosome 17q21.31 microdeletion syndrome is a novel genomic disorder that has originally been identified using high resolution genome analyses in patients with unexplained mental retardation.
Aim: We report the molecular and/or clinical characterisation of 22 individuals with the 17q21.31 microdeletion syndrome.
Results: We estimate the prevalence of the syndrome to be 1 in 16 000 and show that it is highly underdiagnosed. Extensive clinical examination reveals that developmental delay, hypotonia, facial dysmorphisms including a long face, a tubular or pear-shaped nose and a bulbous nasal tip, and a friendly/amiable behaviour are the most characteristic features. Other clinically important features include epilepsy, heart defects and kidney/urologic anomalies. Using high resolution oligonucleotide arrays we narrow the 17q21.31 critical region to a 424 kb genomic segment (chr17: 41046729–41470954, hg17) encompassing at least six genes, among which is the gene encoding microtubule associated protein tau (MAPT). Mutation screening of MAPT in 122 individuals with a phenotype suggestive of 17q21.31 deletion carriers, but who do not carry the recurrent deletion, failed to identify any disease associated variants. In five deletion carriers we identify a <500 bp rearrangement hotspot at the proximal breakpoint contained within an L2 LINE motif and show that in every case examined the parent originating the deletion carries a common 900 kb 17q21.31 inversion polymorphism, indicating that this inversion is a necessary factor for deletion to occur (p<10−5).
Conclusion: Our data establish the 17q21.31 microdeletion syndrome as a clinically and molecularly well recognisable genomic disorder.
Statistics from Altmetric.com
Microdeletion syndromes, such as Prader–Willi syndrome, Williams–Beuren syndrome and velocardiofacial syndrome, were initially clinically described before the underlying causative genomic copy number rearrangement was identified. The introduction of microarray based technology enabled genome profiling in large cohorts of individuals with mental retardation, resulting in the detection of recurrent microdeletions before their clinical description. The chromosome 17q21.31 microdeletion syndrome (Mendelian Inheritance in Man (MIM) 610443) is a new genomic disorder, characterised by a recurrent 500–650 kb deletion involving chromosome 17q21.31.1–3 The recurrent microdeletion was first identified after screening of large heterogeneous cohorts of individuals with mental retardation using high resolution microarray screening technologies.1–3
So far, 14 17q21.31 deletions have been reported in the medical literature,1–8 but for the majority of these cases only limited clinical and molecular data were presented. Further cases and extensive clinical descriptions and molecular studies are needed in order to define the phenotype and genotype of the syndrome.
In all 17q21.31 deletions studied to date the breakpoints map to large clusters of flanking low copy repeats (LCRs), suggesting that the deletions are stimulated by non-allelic homologous recombination (NAHR),9 which is further supported by the identification of the reciprocal duplication in a girl with severe psychomotor developmental delay and dysmorphic craniofacial features.10
We previously estimated the minimal critical region that is recurrently deleted in individuals with the 17q21.31 microdeletion syndrome to a 478 kb region,2 encompassing six genes, including the corticotropin releasing hormone receptor 1 gene (CRHR1) (MIM 122561) and the microtubule associated protein tau gene (MAPT) (MIM 157140). The deletion interval is also the site of a common 900 kb inversion polymorphism, associated with two highly divergent haplotypes designated H1 and H2.11 The H2 lineage, representing the 900 kb inversion polymorphism, is found at a frequency of 20% in the European population.11 So far, the parents of eight affected individuals have been tested and in all cases at least one of the parents carried the H2 haplotype.1–3 Therefore, it has been suggested that the offspring of carriers of the H2 lineage are likely to be predisposed to deletion,12 a phenomenon which has also been described in other microdeletion syndromes, such as Williams–Beuren syndrome, Angelman syndrome, Sotos syndrome,13 and the recently defined 15q13.3 microdeletion syndrome.14
Here we report a collection of 22 individuals with the 17q21.31 microdeletion syndrome. We determine the size of each deletion, allowing detailed delineation of the critical region. We also present the H1/H2 genotypes of the parents, further consolidating the involvement of the H2 haplotype in the deletion. Importantly, based on detailed clinical information of all deletion carriers, the clinical phenotype of the syndrome is characterised and an estimate of the prevalence of the 17q21.31 microdeletion syndrome is obtained.
SUBJECTS AND METHODS
Study subjects
Twenty-two individuals (13 males and 9 females) with a 17q21.31 deletion were included in this study. In three cases the 17q21.31 deletion was suspected, before molecular studies, based on the clinical features of the patient. All 19 other cases had been included in broader cohorts of patients with mental retardation screened for genomic copy number changes. The identification of the 17q21.31 deletion in 11 individuals has previously been reported.1–8 The other cases were identified later using different molecular techniques: multiplex ligation dependent probe amplification (MLPA),15 16 1 Mb resolution bacterial artificial chromosome (BAC) array comparative genomic hybridisation (CGH),6 7 quantitative multiplex polymerase chain reaction (PCR) of short fluorescent fragments (QMPSF),17 fluorescence in situ hybridisation (FISH), segmental duplication BAC microarray (SD array),18 chromosomal microarray analysis (CMA) version V6.0 (Baylor),19 whole genome oligonucleotide array CGH (Agilent Human Genome CGH Microarray Kit 44A and 244A, Agilent Technologies, Santa Clara, California, USA),20 and single nucleotide polymorphism (SNP) microarray (GeneChip Human Mapping 100K, Array Set, Affymetrix, Inc, Santa Clara, California, USA).21 Clinical information and facial photographs were obtained from the referring clinicians. All legal representatives of the patients gave informed consent for the molecular studies and publication of clinical data.
In total 122 individuals (66 males and 56 females) with learning and/or speech and language delay who had a phenotype suggestive of the 17q21.3 microdeletion syndrome, based on facial characteristics and hypotonia, but who did not carry the recurrent deletion, were included in the study for sequencing of the MAPT gene. Microdeletion of 22q11.2 had been excluded previously in 48 patients.
This study was approved by the Medical Ethics Committee of the Radboud University Nijmegen Medical Centre.
Deletion mapping by high resolution microarray
The 17q21.31 deletions that had not been ascertained by a high resolution microarray platform and for which genomic DNA was available were further characterised. For this study, eight cases were tested using a Nsp1 250K SNP array, which contained 262,264 25-mer oligonucleotides. All SNP array experiments were performed according to manufacturer’s protocols (Affymetrix, Inc). Copy number estimates were determined using the CNAG software package (v2.0).22
In order to fine-map deletion breakpoints we used an ultra-high density custom oligonucleotide array (NimbleGen Systems, Madison, Wisconsin, USA). This array consisted of 385 000 isothermal 45–75 mer probes covering six genomic regions, including 121 041 probes specifically targeted to two 300 kb intervals (chr17:40800000–41100000 and chr17:41550000–41850000, hg17), corresponding to the putative breakpoints of the 17q21.31 microdeletion (mean density, one probe per 5.2 bp). Hybridisations were performed as described previously.18 23
Genotyping for H1 and H2, and parent-of-origin analysis
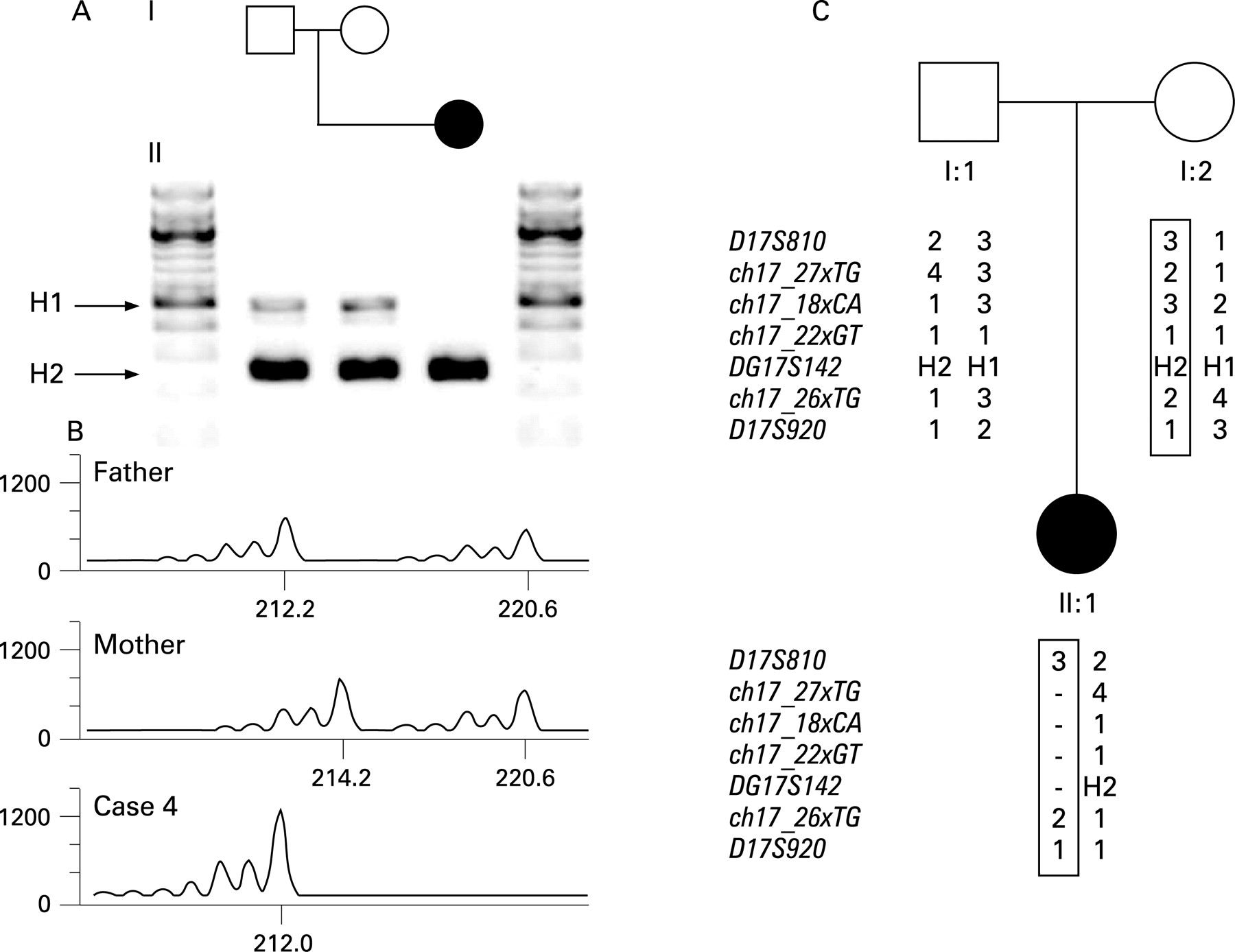
The presence of a 238 bp deletion in intron 9 of the MAPT gene, characteristic for the H2 background,11 24 25 and the dinucleotide marker DG17S142 in intron 9 of the MAPT gene11 were used to genotype individuals with the 17q21.31 deletion and the accompanying parental DNA samples. The 238 bp deletion was determined through visualising by gel electrophoresis. PCR reactions were performed using primer sequences GGAAGACGTTCTCACTGATCTG (sense) and AGGAGTCTGGCTTCAGTCTCTC (antisense) as described previously (supplemental methods).24 The dinucleotide marker DG17S142, four additional variable number tandem repeats (VNTR) inside the deletion interval, and two flanking short tandem repeats D17S810 and D17S920, were used to study the parental origin of the deletions (supplemental methods). All marker analyses were performed according to standard procedures and the size of the peaks were calculated with GeneMapper (v3.7) software (Applied Biosystems, Foster City, California, USA).
Mutation screening of MAPT
One hundred and twenty-two patients who presented with features potentially indicative of a deletion of 17q21.31 were identified from clinical records. Each patient was first tested by MLPA (n = 15) or array CGH using a custom segmental duplication BAC microarray (n = 107),2 and found not to carry the recurrent 17q21.31 deletion. A further seven patients with known deletions of 17q21.311 2 were also selected. Primers targeting all exons and splice sites of the MAPT gene were designed (supplemental methods) and whole genome amplification of genomic DNA (REPLI-g Kit, Qiagen Inc, Valencia, California, USA) performed to yield sufficient quantities of DNA for sequencing. High throughput bidirectional dideoxynucleotide sequencing of PCR amplified gene products was performed (http://genome.wustl.edu/activity/med_seq/protocols.cgi) at the Genome Sequencing Center (Washington University, St Louis, USA) and Department of Human Genetics Nijmegen (RUNMC, Nijmegen, The Netherlands) using standard protocols. PolyPhred,26 and PolyScan27 software were used to generate an automated report of sequence variations by comparison against reference sequences listed in the NCBI (RefSeq) database. Chromatograms were visually inspected for confirmation of non-synonymous sequence variations.
RESULTS
Identification of novel 17q21.31 deletions
The individuals in whom a 17q21.31 deletion was identified were ordered by age at diagnosis, ranging from 10 months in case 1 to 26 years in case 22 (mean age at diagnosis 9 years). The identification of the 17q21.31 deletion in cases 3, 5, 6, 8, 10, 12, 16, 17, 20–22 have previously been reported.1–8 In addition, 11 previously unreported 17q21.31 deletions were ascertained: four novel deletions were found using targeted techniques (MLPA, n = 2; QMPSF, n = 1; FISH, n = 1), two deletions by semi-targeted techniques (SD array, n = 1; CMA V6.0, n = 1), and five deletions were identified using whole genome screening technologies (1 Mb resolution BAC array, n = 1; 100K SNP array, n = 2; Agilent whole genome oligonucleotide array, n = 2). Results of the FISH, MLPA, and QMPSF analyses, and an example of a chromosome 17 profile obtained using whole genome microarray technology, are shown in fig 1. Parental samples were available for 21 patients, and showed that in each case the deletion had arisen de novo.

Deletion mapping by high resolution microarray analysis
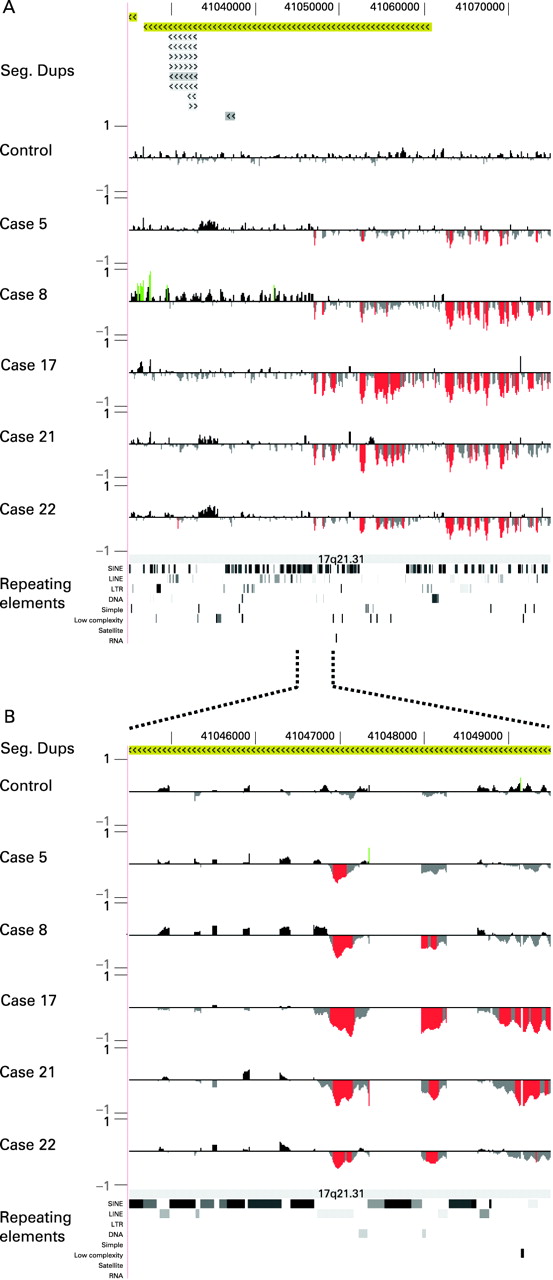
Refinement of the 17q21.31 deletion intervals was conducted using 250K SNP array on DNA samples from eight patients (cases 4, 6, 7, 10, 13–15, 19) and using ultra high density custom oligonucleotide array targeted to the 17q21.31 region in five cases (cases 5, 8, 17, 21, 22). All cases had an overlapping deletion of the 17q21.31 interval.
The SNP array analyses revealed a 458 kb region (chr17:41012856–41470954, hg17) that is recurrently deleted in the eight affected individuals but for which aneuploidy has not been reported in a control cohort of 240 individuals using similar arrays (unpublished results). The proximal breakpoint resided within a 100 kb region in all cases, whereas the distal breakpoint showed more variation. Note that the distal breakpoint is also a site of frequent copy number variation in controls, most likely confounding the mapping and interpretation of this deletion breakpoint.
To define further the breakpoints we characterised the deletion in five different patients using an ultra high density custom oligonucleotide array targeted to the 17q21.31 region, with a mean density of one probe per 5.2 bp. These data show that all five patients have proximal deletion breakpoints that map within an interval of <500-bp (chr17:41046729–41047168, hg17), contained within a single L2 LINE motif (fig 2). Testing of nine control subjects (including carriers of both the H1 and H2 haplotypes) showed that this hybridisation signature was specific to deletion patients and does not represent a copy number variation. In contrast, we were unable to identify a deletion signature at the distal breakpoint which was specific to deletion carriers. On the basis of the location of the L2 LINE element at the proximal side and the copy number variation identified in 240 normal controls at the distal side, we refined the critical region to an 424 kb genomic interval (chr17:41046729–41470954, hg17), relative to the H1 lineage.
Genotyping
We performed genotyping of the H1 and H2 haplotypes using a dinucleotide marker (DG17S142) in intron nine of MAPT and a characteristic 238 bp deletion in the same intron.11 24 25 In all parents tested (n = 42) at least one of the parents carried the H2 inversion and seven parents were homozygous for the H2 inversion. Genotyping of D17S810, DG17S142 and D17S920 and four polymorphic loci, designed in the deleted region, showed that the parent-of-origin carried the H2 inversion polymorphism (table 1, fig 3). In total, eight deletions were of maternal origin and 12 were of paternal origin, whereas in two cases marker analysis could not distinguish between the parental chromosomes.
Mutation screening of MAPT
A full list of nucleotide changes identified in the 122 non-deletion patients tested is shown in supplemental table 1. Two novel non-synonymous mutations were identified in MAPT: (1) an identical heterozygous G>A transition at hg17 position chr17:41443588, resulting in the replacement of valine with isoleucine in an alternatively spliced exon, was detected in two unrelated patients; (2) a heterozygous G>A transition at hg17 position chr17:41457221, resulting in the replacement of glycine with arginine. Screening of seven 17q21.31 deletion patients did not identify any novel variants.
Clinical details of the study subjects
The clinical features of the patients with a 17q21.31 deletion are listed in table 2 while extensive detailed descriptions are provided in the supplemental notes. A summary of the 22 patients is given below.
In the majority of cases (82%) the pregnancy was uneventful. In case 2, however, the pregnancy was characterised by a placental abruption at 7 weeks of gestation, and in cases 9 and 21, the pregnancy was complicated by intrauterine growth retardation. Low birth weight (<3rd centile) was noted in six (27%) individuals. Other birth measurements were within the normal range, although in case 9 microcephaly was noted, and cases 3 and 14 were small for gestational age. Short stature (<3rd centile) was present in four patients (18%). Case 19 was investigated for short stature at 11 years of age, with a borderline low growth hormone measurement on provocation testing.
In all patients global psychomotor developmental delay was noted from an early age. The level of developmental delay varied significantly and was estimated from mild to severe. For example, in case 17 the early motor milestones were delayed, but she started to walk at 19 months, whereas case 10 did not start walking before the age of 4 years. Eleven patients did not have any words before 3 years of age. The speech and language development of case 17 were particularly affected. She did not achieve two-three word sentences until age 6. Case 16 communicated primarily with gestures at 6 years of age, while case 10 showed no understandable speech during the investigation at the age of 6 years 2 months.
Hypotonia with poor sucking and slow feeding was evident in the neonatal period and during childhood in all but one. In at least six patients feeding difficulties required hospitalisation and/or nasogastric tube feeding in the neonatal period.
A history of epilepsy was noted in 50% of the cases. Generalised seizures were present in eight cases (36%). In case 4 unilateral clonic seizures and hypotonia were observed 48 h after birth. A parieto-occipital haemorrhagic infarction on the left side with bilateral ventricular haemorrhage was diagnosed and a large thrombus was identified in the terminal portion of the aorta.
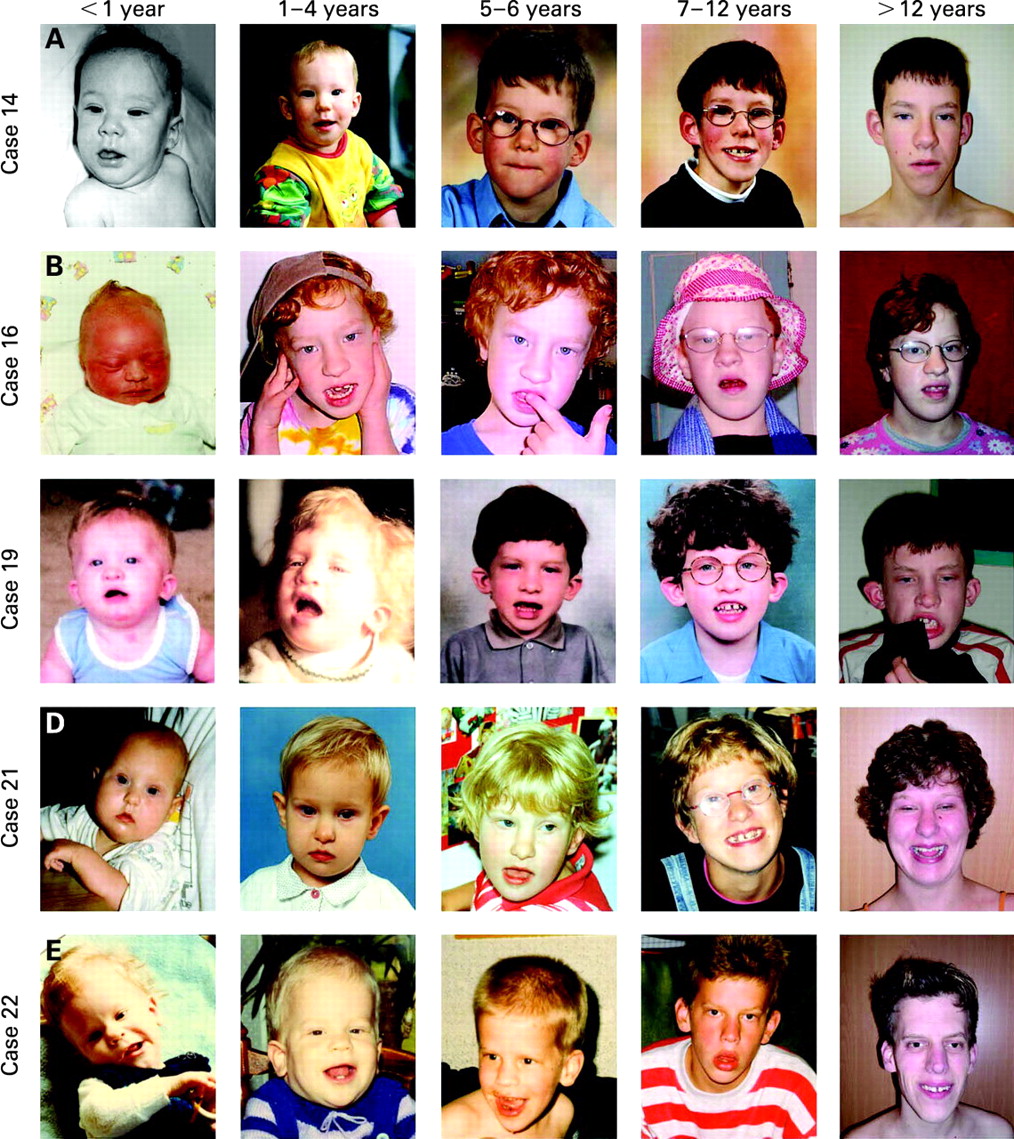
Dysmorphic craniofacial features present in more than half of the individuals included a high/broad forehead, long face, upward slanting palpebral fissures, epicanthic folds, an abnormally formed nose (either “tubular” or “pear” shaped), bulbous nasal tip, large prominent ears, and everted lower lip and, in addition, abnormal hair pigmentation and texture was observed in 12 individuals (55%). The nose can have a high nasal bridge, a broad nasal root, long columella, hypoplastic and/or thick alae nasi. Ophthalmological evaluation showed strabismus in 45% of patients and hypermetropia was present in eight cases (36%). Facial photographs of 20 individuals are provided in fig 4. Facial photographs at different ages, spanning a significant period of time, could be obtained for five individuals (fig 5). The facial characteristics of the patients change with age. In infancy the facial gestalt is mostly characterised by the hypotonia of the face with an open mouth appearance. However, with increasing age there is elongation of the face and broadening of the chin and also the “tubular” or “pear” shape form of the nose becomes more pronounced.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Slender long fingers were reported in 61% of the patients. In addition, hypoplasia of the hand muscles was described in multiple patients (29%) as well as slender lower limbs (41%), dislocation of the hip(s) (27%), and positional deformities of the feet (27%). Case 13 had in-turned feet with significant pronation and had been fitted with an ankle–foot orthosis. He had progressive contractures in his wrists and ankles, and bilateral ulnar deviation of his wrists and deviation of his toes.
Septal heart defects, both atrial septal defects (ASD) and ventricular septal defects (VSD), were found in six cases (27%). Kidney and urologic anomalies were found in seven cases (32%). The latter included vesicoureteric reflux, hydronephrosis, right sided pyelectasis, and a duplex renal system. In case 6 pyelonephritis and bilateral vesicoureteric reflux grade II was detected. In case 14 a renal ultrasound at the age of 5 months showed left hydronephrosis due to a primary pelvi-ureteric junction stenosis. Cryptorchidism was reported in seven out of nine male patients (78%) and deformity of the spine requiring treatment was present in eight patients (36%). The spine anomalies mostly included scoliosis, but lordosis and kyphosis were also reported. In case 13 the scoliosis was progressing rapidly. A brace was required and an x ray suggested that he might be suffering from a mild degree of restrictive lung disease. Magnetic resonance imaging of the spine showed a compression of the upper cervical spinal cord, due to developmental anomaly in the craniocervical junction and upper cervical canal. Imaging of the spine in case 19 showed fusion of the C4/5 vertebrae and two small syringes in the thoracic cord.
In the vast majority of the patients (89%) the behaviour was described as friendly, amiable and cooperative with or without frequent laughing. In contrast to this general pattern, the attention span of case 7 was very short, his interaction with people limited, and he had temper tantrums. Additionally, case 15 showed behaviour problems including hyperactivity alternating with introspection, bad humour, and difficult interaction with other children.
DISCUSSION
The 17q21.31 microdeletion syndrome is a new mental retardation syndrome.1–3 We performed an extensive clinical and molecular characterisation of 22 patients. In the majority of cases the microdeletion was identified after microarray based genomic copy number profiling of large heterogeneous cohorts of individuals with mental retardation. In only three cases was the 17q21.31 deletion suspected, based on the clinical features of the patient (cases 7, 8, 18). The remaining 19 cases had been included in broader cohorts including a total of 2978 patients with mental retardation. Therefore, the prevalence can be estimated to be in the order of 0.64% (95% confidence interval 0.35% to 0.93%) of the individuals with unexplained mental retardation. Mental retardation occurs in approximately 2–3% of the general population,28 29 in approximately 50% of which a diagnosis cannot be made.30 Therefore, we estimate the prevalence of the 17q21.31 deletion syndrome as approximately 1 in 16 000 individuals. For example, for the Netherlands, this implies that ∼12 affected individuals are born each year. So far, only a total of four cases from the Netherlands have been reported, indicating that the 17q21.31 deletion syndrome in individuals with mental retardation is currently highly underdiagnosed.
The 17q21.31 region contains multiple copy number variations (CNVs) that are also found in the general population (http://projects.tcag.ca/variation/), complicating the definition of the precise deletion breakpoints. Based on 250K SNP array analyses we could delineate a minimal 424 kb critical region (41046729–41470954 Mb, hg17) that is recurrently deleted in patients, but not in controls. Further refinement of five deletions using the ultra high density oligonucleotide arrays (mean density, one probe per 5.2 bp) revealed that the proximal breakpoint in all five tested patients is contained within an interval of <500 bp within an L2 LINE motif, representing a possible hotspot for NAHR. Even with the ultra high density oligonucleotide arrays it is not possible to ascertain the distal breakpoint accurately. The deletion seems to extend more distally in some patients tested, although this might reflect the extreme copy number polymorphism at the distal side, rather than variation in the breakpoints. Future deep sequencing unravelling the H2 sequence and a putative common distal breakpoint, which is currently unknown, will be of major importance to prove NAHR as the underlying mechanism.
The 424 kb critical region encompasses at least six genes, C17orf69, CRHR1 (MIM 122561), IMP5 (MIM 608284), MAPT (MIM 157140), STH (MIM 607067), and KIAA1267. Haploinsufficiency of one or more of these genes might underlie the phenotype seen in the 17q21.31 deletion syndrome. The MAPT is of particular interest as the gene is highly expressed in brain and is involved in several neurodegenerative diseases, such as frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17),31 32 progressive supranuclear palsy,33 and corticobasal degeneration and Alzheimer’s disease.25 Therefore, we performed mutation analysis of the entire coding sequence of the MAPT (MIM 157140) gene in 122 patients targeted for testing on the basis of clinical features but lacking the characteristic deletion.
Two non-synonymous mutations of uncertain significance were found in this cohort. The first of these, resulting in the replacement of valine with isoleucine, was identified in two unrelated patients. Although not previously reported, the presence of this same variant in two unrelated individuals suggests that it likely represents a rare polymorphism. Furthermore, the amino acids valine and isoleucine are structurally and biochemically quite similar, suggesting that this substitution is unlikely to significantly affect protein structure. The second variant identified results in the replacement of glycine with arginine at amino acid 389. This same amino acid variant has been previously reported in two families with Pick’s disease (frontotemporal dementia, MIM 172700).34 35 However, this variant only has mild effects on MAPT function and in neither of these reports did the mutation segregate with the neurological phenotype, suggesting that this may also represent a rare polymorphism. Although our extensive efforts to identify pathogenic mutations in MAPT did not uncover any variants that seem likely to be responsible for the phenotype, no firm conclusions can be made, because parental DNA samples were unavailable for further testing. These data suggest that the 17q21.31 microdeletion syndrome is not caused solely by haploinsufficiency of MAPT, although single exon deletions could have been missed in our analyses. We speculate that the phenotype of the 17q21.31 deletion syndrome instead results from haploinsufficiency for one or more other elements within the critical region, perhaps representing a contiguous gene syndrome. This is in contrast to some other genomic disorders in which the associated phenotypes largely or completely result from haploinsufficiency for single genes—for example, the RAI1 gene in Smith–Magenis syndrome,36 and the UBE3A gene in Angelman syndrome.37
The 17q21.31 genomic interval contains a common 900 kb inversion polymorphism, resulting in a haplotype block with two highly divergent haplotypes designated H1 and H2.11 Genotyping the parents with respect to the H1/H2 lineage showed that in each trio tested the parent originating the deleted chromosome 17 carries at least one H2 chromosome, which is significantly different from the ∼20% frequency of the inversion in the European population reported by Stefansson et al11 (p<10−5, Pearson’s χ2 test). In total, eight deletions were of maternal origin and 12 were of paternal origin, indicating that there is no significant bias for parental origin of the deletion. The H2 haplotype results in a genomic structure with directly oriented LCR subunits that can undergo a deletion rearrangement via NAHR,38 which suggests that the inversion found in all parents of origin may be a necessary factor for the deletion to occur. Although the H2 allele is a risk factor, the frequency of de novo 17q21.31 microdeletions in carriers of the H2 inversion is low; therefore, other as yet poorly understood factors are likely to be important in the generation of the deletion. Moreover, all deletions detected to date are sporadic. Therefore, assuming the H2 allele is requisite for the deletion, based on current knowledge, the occurrence risk for the 17q21.31 deletion in a carrier of the H2 inversion polymorphism might be considered to be in the order of 1/3200 in European populations. However, from a genetic counselling perspective, the recurrence risk in a family of an affected individual could be higher, because of other factors such as germline mosaicism.
Our analysis of 22 patients with the 17q21.31 deletion syndrome shows a clinically recognisable phenotype. Common features present in more than 50% of the patients that should prompt consideration of this diagnosis include developmental delay, childhood hypotonia, abnormal hair colour/texture, high/broad forehead, long face, upward slanting palpebral fissures, epicanthal folds, tubular or pear shaped nose, bulbous nasal tip, large/prominent ears, slender/long fingers, cryptorchidism, and a friendly/amiable behaviour. Other common features that need special medical attention are epilepsy, hypermetropia, pectus excavatum, congenital heart defects (VSD/ASD), kidney and urologic anomalies, dislocation of the hip(s), positional deformities of the feet, and spinal deformities. Hypotonia of the face is most obvious in infancy, with an open mouth appearance, everted lower lip and a protruding tongue. The typical nose and other facial characteristics can be observed from birth. However, with increasing age, there is a change in phenotype, which is also seen in other mental retardation syndromes, such as Mowat–Wilson syndrome,39 Noonan syndrome,40 and Williams–Beuren syndrome.41
In conclusion, the molecular and/or clinical characterisation of 22 individuals with the 17q21.31 microdeletion syndrome defines the phenotypic features associated with this novel syndrome and provides further insight into the critical region and rearrangement hotspot. Our data further support the hypothesis that the common 17q21.31 inversion polymorphism in the parent-of-origin is a necessary factor for the deletion to occur.
Acknowledgments
We thank all the parents and children who have participated in this study. We would also like to acknowledge Dr Regina Regan and Mrs Cheryl Guiver (Oxford Genetics Knowledge Park), and Cindy Skinner for their assistance.
REFERENCES
Supplementary materials
web only appendices 45/11/710
Files in this Data Supplement:
Footnotes
Competing interests: None.
Funding: This work was supported by grants from the AnEUploidy project (LSHG-CT-2006-037627) supported by the European commission under FP6, and supplemental grants from the Netherlands Organisation for Health Research and Development (ZonMW 907-00-058, ZonMW 917-86-319 to BBAdV, ZonMW 920-03-338 to DAK, ZonMW 912-04-047 to HGB and JAV), Hersenstichting Nederland (BBAdV), South Carolina Department of Disabilities and Special Needs (CES) and the National Institutes of Health (NIH) (EEE, HD043569). EEE is an Investigator of the Howard Hughes Medical Institute.
Patient consent: Parental consent obtained
Linked Articles
- Correction