Article Text

Abstract
Cardio-facio-cutaneous (CFC) syndrome, Noonan syndrome (NS), and Costello syndrome (CS) are clinically related developmental disorders that have been recently linked to mutations in the RAS/MEK/ERK signalling pathway. This study was a mutation analysis of the KRAS, BRAF, MEK1 and MEK2 genes in a total of 130 patients (40 patients with a clinical diagnosis of CFC, 20 patients without HRAS mutations from the French Costello family support group, and 70 patients with NS without PTPN11 or SOS1 mutations). BRAF mutations were found in 14/40 (35%) patients with CFC and 8/20 (40%) HRAS-negative patients with CS. KRAS mutations were found in 1/40 (2.5%) patients with CFC, 2/20 (10%) HRAS-negative patients with CS and 4/70 patients with NS (5.7%). MEK1 mutations were found in 4/40 patients with CFC (10%), 4/20 (20%) HRAS-negative patients with CS and 3/70 (4.3%) patients with NS, and MEK2 mutations in 4/40 (10%) patients with CFC. Analysis of the major phenotypic features suggests significant clinical overlap between CS and CFC. The phenotype associated with MEK mutations seems less severe, and is compatible with normal mental development. Features considered distinctive for CS were also found to be associated with BRAF or MEK mutations. Because of its particular cancer risk, the term “Costello syndrome” should only be used for patients with proven HRAS mutation. These results confirm that KRAS is a minor contributor to NS and show that MEK is involved in some cases of NS, demonstrating a phenotypic continuum between the clinical entities. Although some associated features appear to be characteristic of a specific gene, no simple rule exists to distinguish NS from CFC easily.
- CFC syndrome
- Costello syndrome
- Noonan syndrome
- KRAS
- BRAF
- HRAS
- MEK1
- MEK2
Statistics from Altmetric.com
Since its original description by Reynolds et al,1 cardio-facio-cutaneous (CFC) syndrome has been reported in about 60 patients, allowing precise elucidation of its phenotype.2–5 The developmental anomalies in CFC include congenital heart defects (CHD), ectodermal anomalies, and short stature. The degree of mental retardation is variable, usually moderate to severe. Affected individuals also present a characteristic facial appearance with a high forehead, bi-temporal constriction, down-slanting palpebral fissures, short nose with depressed nasal bridge, and relative macrocephaly. The CFC phenotype is reminiscent of Noonan syndrome (NS) and Costello syndrome (CS) and differential diagnosis can be difficult, particularly in infancy. Diagnostic indexes have been proposed by Grebe and Clericuzio6 and Kavamura et al.7 Some clinical signs are useful to differentiate the three entities clinically. Sparse hair and eyebrows, follicular hyperkeratosis and palmoplantar hyperkeratosis characterise CFC, whereas cutis laxa, diffuse skin hyperpigmentation, papillomata, ulnar deviation of the hands and nail dystrophy are hallmarks of CS. Qualitatively, facial dysmorphology is similar in NS, CS and CFC, but compared with patients with NS, the face of patients with CFC is wider. The mouth of patients with CS is also wider with thick lips, and coarsening of face is typical in both CS and CFC. CHD in CFC are remarkably similar to those noted in NS and CS. The incidence of CHD and hypertrophic cardiomyopathy are comparable in CFC and CS. Severe cardiac arrhythmias occur in a third of patients with CS, whereas they are rare in NS and CFC. In infancy, feeding problems and failure to thrive are more frequent and severe in CFC and CS than in NS. CS and CFC are associated with more severe developmental delay than NS. CS is associated with a greatly increased risk of malignancy, notably rhabdomyosarcoma. The incidence of CFC is unknown. All cases reported to date have been sporadic.
In 2001, Tartaglia et al discovered that activating mutations of PTPN11 cause 40–50% of cases of NS.8 As PTPN11 encodes SHP2, a non-receptor tyrosine phosphatase involved in RAS pathway activation, genes encoding RAS/MAPK components have systematically been screened. Activating mutations of HRAS were found in roughly 85% of patients with a clinical diagnosis of CS.9–12 Patients with CFC harbour activating missense mutations in KRAS,13 BRAF,13 14 MEK1 and MEK2.14 KRAS was also shown to cause a small subset of NS cases.15 16 About 10% of patients with NS carry activating mutations in SOS1, a RAS-activating molecule of the guanosine exchange factor (GEF) family.17 18 These proteins are part of the RAS/MAPK signalling pathway, which is involved in many biological processes and plays crucial roles during embryonic development.19 Somatic mutation and/or increased transcription of the genes encoding these proteins are a common feature in tumour progression.
The aim of this study was to screen the genes causing CFC syndrome in three cohorts of patients referred with (1) a clinical diagnosis of CFC, (2) a clinical diagnosis of CS but no HRAS mutation, or (3) with a diagnosis of NS without PTPN11 or SOS1 mutation, to establish the pattern and frequency of mutations in these diseases, to delineate the overlap between these clinically related syndromes and to investigate possible genotype–phenotype correlations.
PATIENTS AND METHODS
Patients
Our original cohort comprised 53 patients with CFC syndrome; 13 patients, previously reported,13 20 are not included in this paper. The study thus comprised 40 new patients with a clinical diagnosis of CFC, 20 patients with a clinical diagnosis of CS but no HRAS mutation, and 70 patients with NS but no PTPN11 or SOS1 mutation.
Patients with CFC and NS were referred for molecular testing to our laboratory by a network of geneticists from France, Belgium and Switzerland. Patients with CS were found through the French Costello Syndrome Association. A diagnosis of CS had been proposed at some time in all these patients by a clinical geneticist. It was usually based on severe developmental delay, failure to thrive and/or skin anomalies, and was the referral diagnosis for all patients within this group. This group is clinically more heterogeneous, mixing patients with truly convincing CS and patients who would probably have been diagnosed as CFC by trained dysmorphologists, but who were still carrying a diagnosis of CS and remained in the Costello Support Group. As these uncertainties in diagnosis may reflect a general difficulty in clinical differentiation between CS and CFC, we decided to keep the diagnoses of referral. Pictures of the patients were collected, and a questionnaire containing 72 clinical items about neonatal data, characteristic facial features, heart defects, skin abnormalities, growth retardation, developmental delay or mental retardation, and occurrence of solid tumour or leukaemia was used to collect clinical data. Informed signed consent for genetic investigation was obtained from all patients or their parents.
All cases of CFC and CS were apparently sporadic, with clinically and developmentally normal parents. The same statement applied to patients with NS, although it is known in this syndrome that expressivity of a mutation in a carrier may be sufficiently mild to remain clinically unsuspected (at least for patients carrying mutations in PTPN11).
Molecular analysis
DNA samples were obtained from peripheral leucocytes. In one patient, DNA from cultured fibroblasts was also tested. Mutation screening was performed by direct bidirectional sequencing of exons and their flanking intron–exon boundaries. The entire coding region of KRAS, BRAF, MEK1, MEK2, PTPN11 and HRAS was tested in all patients. Primers and PCR conditions are available on request.
The PCR products were sequenced (Big Dye Terminator Cycle Sequencing Ready Reaction Kit; (Applied Biosystems, Foster City, California, USA), and reaction products run on an automated capillary sequencer (ABI 3100 Genetic Analyzer, Applied Biosystems). Sequences were aligned using Seqscape analysis software (Applied Biosystems) and compared with the reference sequences for genomic DNA and mRNA. GenBank accession number for genomic and mRNA reference sequences, respectively, are as follows: KRAS NC_000012 and NM_033360 (isoform a) or NM_004985 (isoform b), BRAF NC_000007 and NM_004333, MEK1 NC_000015 and NM_002755, MEK2 NC_000019 and NM_030662, PTPN11 NC_000012 and NM_002834, HRAS NC_000011 and NM_176795.
The Catalogue for somatic mutations in cancer (http://www.sanger.ac.uk/genetics/CGP/cosmic) was used to check for previous implication of the mutations in cancer. Presence of single-nucleotide polymorphisms was ascertained using the Ensembl genome browser (http://www.ensembl.genome.org). Interspecies alignments and prediction of functional effects of amino acid substitutions on the function and structure of proteins were achieved using PolyPhen. (http://genetics.bwh.harvard.edu/).
RESULTS
Mutation screening
In total, 12 BRAF mutations including 5 unreported mutations (T241P, Q262R, G464R, E501V, N581K) were identified in 22 patients (fig 1A). All patients had CFC (n = 14) or CS (n = 8). There were 14/22 (64%) patients with a mutation in exon 6, with a hot spot on Q257. A mutation of exon 6 was found in seven of the eight patients with CS, whereas mutations associated with CFC tended to be more evenly distributed (fig 1A). All mutations occurred in exons previously shown to harbour CFC mutations. No mutation was found in exons 13 or 16.

Four MEK1 and 4 MEK2 mutations, including 3 novel mutations for MEK1 (E44G, T55P, D67N), and 3 novel mutations for MEK2 (L46_E55del, K61T, A62P) were identified in 15 patients with CFC (n = 8), CS (n = 4), or NS (n = 3) (fig 1B, 1C). Three patients with NS had a novel mutation in the exon 2 of MEK1. All mutations were found in exons already identified as mutational hot spots.
Five KRAS mutations, including two unreported mutations (K5E, G12S) were identified in seven patients with CFC (n = 1), CS (n = 2) or NS (n = 4) (fig 1D). All mutations occurred in exons 1, 2 and 4b. No PTPN11 mutation was found in patients with CFC or a CS, and none of the patients referred with CFC had a HRAS mutation.
Altogether, a mutation of one of the tested genes was found in 23/40 (57%) patients with CFC syndrome, in 14/20 (70%) patients with CS and in 7/70 (10%) patients referred for NS and who were negative for PTPN11 and SOS1 mutation (table 1). All identified mutations except one were missense mutations, and all kept the reading frame open.
All cases with a mutation were considered by the referring clinician to be sporadic. The presence of the mutation could be investigated in both parents for 25 cases (12 with a BRAF mutation, 6 with a MEK1 mutation, 4 with a MEK2 mutation, and 3 with a KRAS mutation) and in the mother only for 4 cases (3 with a BRAF mutation, 1 with a KRAS mutation). The mutation was not found in the parents, with exception of one patients with NS, who had a novel MEK1 mutation (E44G) inherited from her asymptomatic mother. No BRAF mutation was found in patients with NS.
Overall, 14 novel mutations were found in 17 patients. De novo occurrence could be confirmed for six mutations (eight patients), by testing the parents’ DNA (table 2). This favours a causative effect of these mutations. Pathogenicity of the MEK1 alteration found in a NS patient and her clinically unaffected mother cannot be solved so easily. The substitution has not been previously reported and we did not find it in a series of 200 normal subjects with similar ethnic background. This substitution may represent a rare polymorphism or an incompletely penetrant mutation. In the cases for which parental DNA was not available, pathogenicity was considered likely, as these alterations were not identified in 200 controls and have not been reported as polymorphisms. In most cases, the affected amino acids were highly evolutionarily conserved and predicted to be deleterious (table 2).
Most germline mutations identified in our patients are distinct from the somatic mutations present in cancers. Four patients (aged 1.5, 4.5, 8.7 and 14.3 years at the last examination) carry mutations previously reported in tumours (BRAF G464R, KRAS G12S and KRAS V14I in two patients) (fig 1). The G12S mutation in KRAS was also present in fibroblasts of the second child (now aged 8.7 years). The median age at clinical diagnosis was 1, 1.7, and 2 years and the median age at molecular diagnosis was 4.7, 7.7 and 8.7 years for the patients with BRAF, MEK and KRAS mutations, respectively. None of these children has developed cancer to date.
Clinical description
Because of the probable genetic heterogeneity of patients with no identified mutations, we did not perform comparisons of patients with and without mutations. We compared the phenotypes of patients according to the mutated gene and the initial clinical diagnosis (CFC or CS). Clinical data of patients with CFC were then compared with those of the series of Kavamura et al,7 which was a study of 54 patients with CFC before molecular diagnosis. Finally, patients with CS without HRAS mutation were compared with the patients with CS with HRAS mutations described by Kerr et al10 (table 3).
All our patients with CFC have the classic dysmorphism (hypertelorism, downslanting palpebral fissures, ptosis, high forehead with bitemporal constriction, short neck). Hair anomalies were found in 95%, and sparse or absent eyebrows in 78%. CHD was recorded in 77%. These features are in agreement with the series of Kavamura et al (table 3). However, in contrast with that study, our patients have a more severe neurological presentation, with hypotonia in 68% (vs 28%, p⩽0.01), speech delay in 95% (vs 46%, p⩽0.001), and mental retardation in 100% (vs 91%, NS). In our series, growth retardation was postnatal, with a median birth weight of 3110 g for a mean gestational age of 37 weeks. Short stature (<−2SD) was less frequent in our patients than in those reported by Kavamura et al (56% vs 78%) although this difference was not significant.
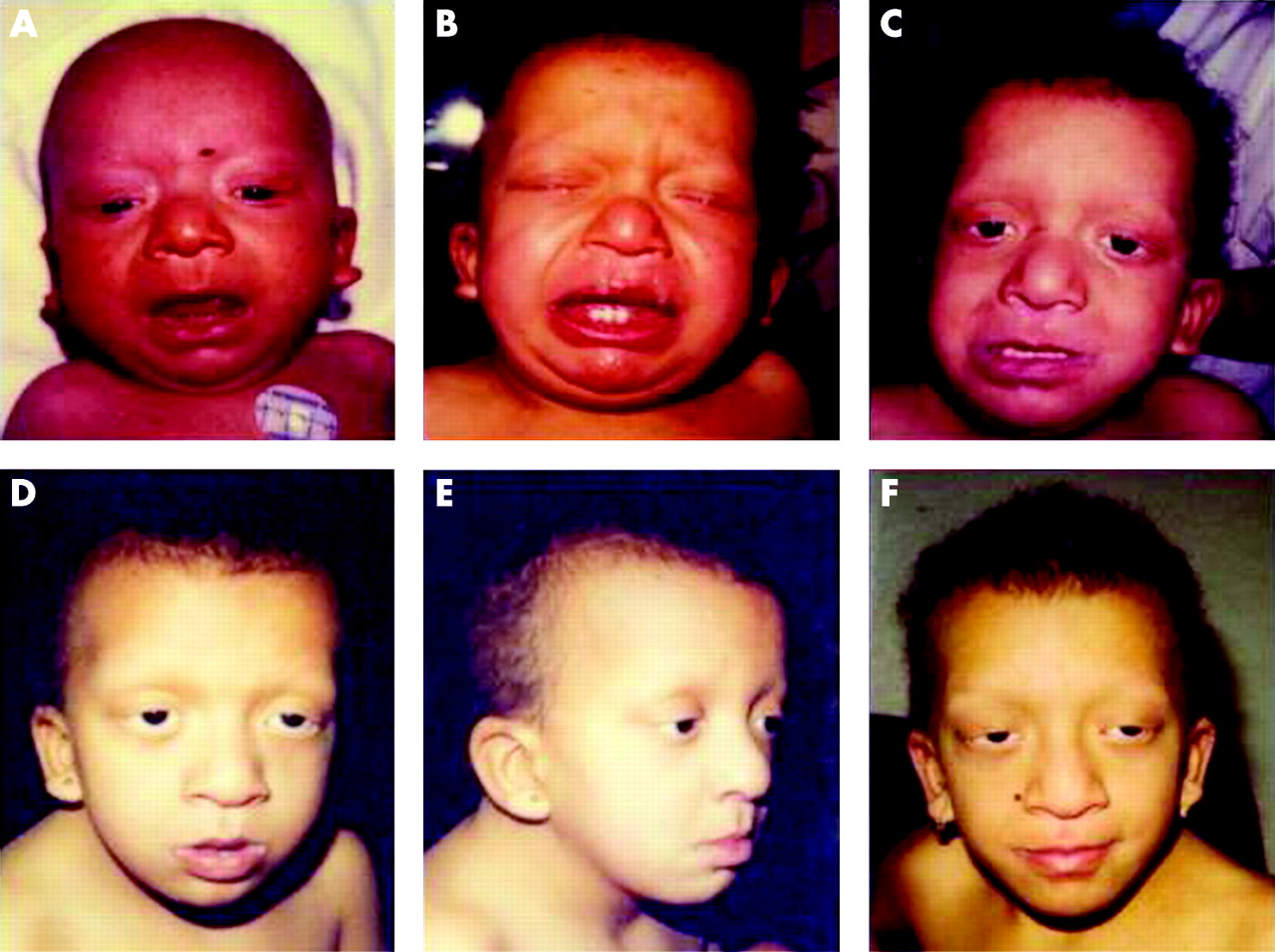
The dysmorphic features observed in patients with CS are those usually considered typical for this CFC syndrome also. These patients show a similar incidence of heart defects and failure to thrive to French and British patients with CS with HRAS mutation.10 However, our patients with CS are younger than those reported by Kerr et al10 (median 6 years vs 9 years) and six patients were diagnosed with CS before the age of 2 years. They present features overlapping with CFC, notably sparse or absent eyebrows in 92% of cases, in contrast to patients with CS and HRAS mutations, who have normal eyebrows. Moreover, none of our patients with CS presents papillomata, one of the more distinctive features of CS. Therefore, it is likely that some patients are actually misdiagnosed CFC cases. However, our patients with CS have a more severe phenotype than those with CFC. They present more hypotonia, failure to thrive and growth retardation are more marked in infancy, and large mouth, thick lips and coarse facies are more frequent. Developmental delay is more marked; age at first steps was 3.0 years versus 2.1 years for patients with CFC. Most of them present deep palmar creases and skin hyperlaxity, which were often considered characteristic of CS, and probably have contributed to their clinical diagnosis (fig 2).

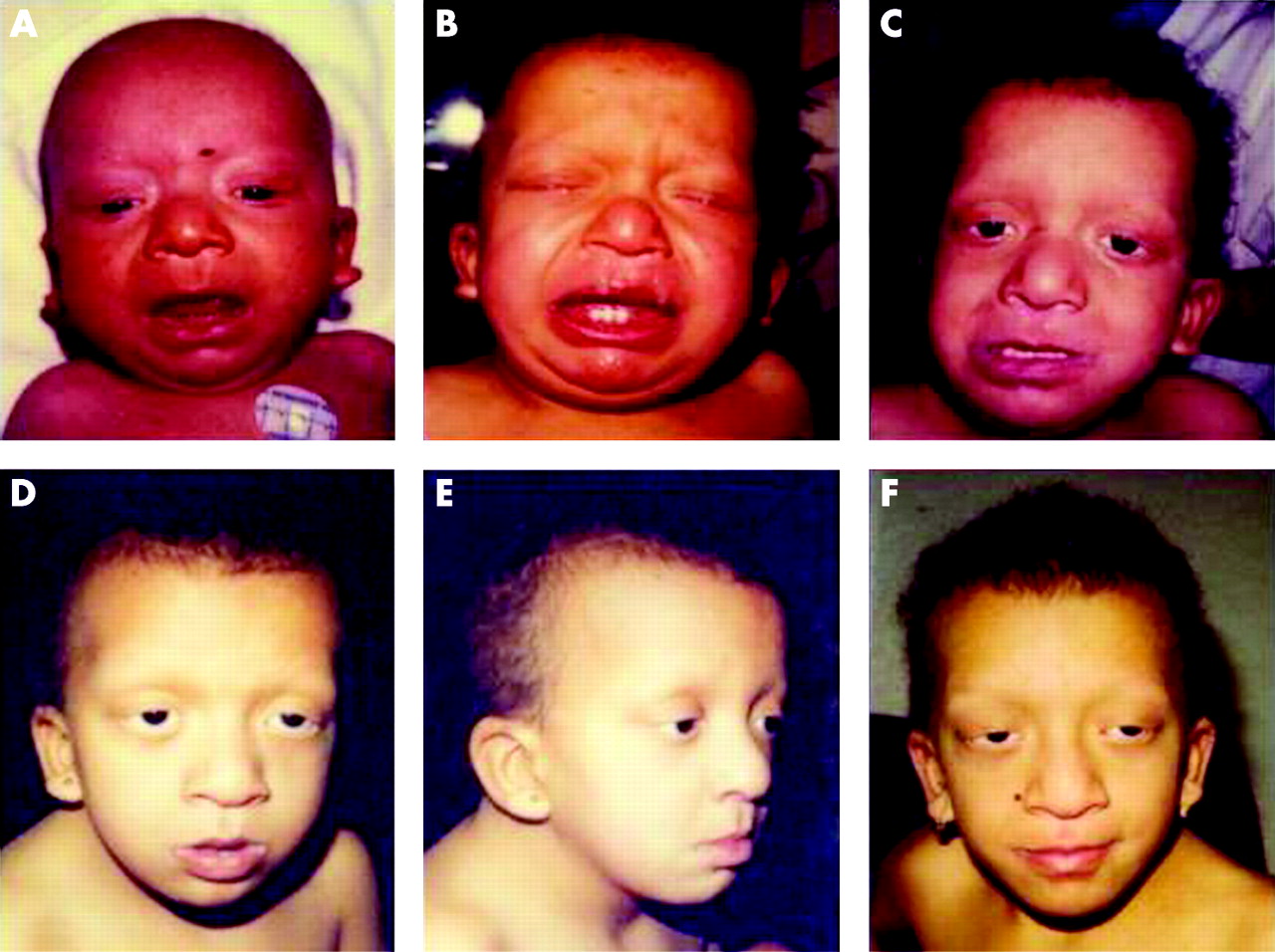
In general, patients with a MEK1 or MEK2 mutation present with a milder phenotype than those with a BRAF mutation. Heart defect is less frequent (43% vs 90%, p⩽0.001) (table 3). Motor delay tends to be milder (median age of walking 2 years vs 2.5 years for BRAF and 2.7 years for KRAS) and two patients have no mental retardation. Dysmorphism less commonly includes hypertelorism (p⩽0.05) or sparse hair (p⩽0.01). Skin anomalies are similar to those reported with CS: coarse facies (9/12), deep palmar/plantar creases (7/10), redundant skin folds on hands and feet (5/11) and hyperextensible joints (8/11). A recurrent novel mutation (D67N) was found in three patients (proven to be de novo in two). One of these patients has CFC syndrome. He has relative macrocephaly, wide face, temporal constriction, curly hair, sparse brows and lashes, pulmonary valve stenosis, failure to thrive and developmental delay. The second, aged 12 years, has typical NS: short stature, triangular face without temporal constriction, non-curly hair, ptosis, almost absent eyebrows and borderline intelligence with hyperactivity–attention deficit disorders. He is able to have normal schooling with extra help. The third, diagnosed as mild NS, has short stature, hypertelorism, wide face without temporal constriction, normal brows and non-curly hair, no failure to thrive, pulmonary valve stenosis, and normal psychomotor development at 6 years of age. The evolution of the phenotype with age must be taken into account, as illustrated by one of our patients with CFC who had a NS phenotype in infancy (fig 3).
{kind=link}
{kind=link}
{kind=link}
The four patients having NS with a KRAS mutation were considered to have the typical NS gestalt, notably the triangular shape of the face, and absence of major skin involvement. They are nevertheless at the severe end of the NS spectrum: marked developmental delay, short stature, heart defects (two pulmonary valve stenosis, one mitral valve defect associated with hypertrophic cardiomyopathy, one hypertrophic cardiomyopathy). Three of the four have failure to thrive. Sparse hair (2/4) and eyebrows (1/4) indicate a clinical overlap with CFC in two of these patients.
DISCUSSION
Our results confirm the high proportion of patients with BRAF mutations in CFC, illustrate the clinical overlap between the phenotype of patients with HRAS mutations compared with KRAS and their downstream effectors, and suggest, to our knowledge for the first time, the implication of MEK1 in NS.
The mutation frequency observed in our series of 40 patients with CFC (57%) is in accordance with the data from Narumi et al20 (35/56; 62%), but is clearly lower than the mutation rate reported by Rodriguez-Viciana14 (21/23; 91%). This difference is mainly due to a higher mutation rate of BRAF in the latter series (78% vs 35%) and is probably caused by more stringent clinical criteria, as patients with a BRAF mutation are, as a whole, more typical than those with mutations in the other genes.
A mutation in BRAF, KRAS or MEK1 was found in 70% of patients clinically diagnosed as CS but without HRAS mutation, whereas HRAS mutation was not found in patients with a clinical diagnosis of CFC. This observation, together with the clinical presentation of these patients, suggest that CFC is clinically closer to CS than previously appreciated, to a point that distinction in a single individual may be impossible, at least in infants and young children. Indeed, early manifestations (such as deep palmar creases or severe failure to thrive), which were once thought to be “specific” for CS, are in fact present with or without HRAS mutation. As patients with HRAS mutations age, some clinical features (arrhythmia, multiple papillomas, facial coarseness, preservation of eyebrows) allow easier distinction between CFC and CS. Our data suggest that mutations within the cysteine-rich domain of BRAF could be associated with a phenotype closer to CS, whereas mutations in the protein kinase domain result in a phenotype more typical for CFC. However, the small number of patients meant this did not reach significance.
Patients with KRAS mutations presented the most variable phenotype, confirming the experience of Zenker et al.21 One of these was diagnosed with CFC, four with NS, and two with CS. The phenotype was generally severe, with hypotonia, short stature, and heart defect in all cases and failure to thrive in 6/7 patients. One of our patients (with V14I mutation) has no mental retardation. He presented developmental delay in infancy, with first steps at 2.1 years and first words at 2.3 years. He now has normal schooling at 14 years of age. This confirms a recent observation22 of high intelligence in a patient with KRAS-associated familial NS. However, this latter patient had a mutation restricted to isoform a, which is not the case in our patient. We confirm that patients with KRAS mutation may have hypotrichosis but not hyperkeratosis.
We also confirm the implication of KRAS in NS. We identified KRAS mutation in 5% of PTPN11-negative and SOS1-negative patients (4/70), a proportion similar to the findings of Schubbert et al (5/175 PTPN11-negative patients with NS).15 Mutation V14I is recurrently associated with NS, indicating a possible genotype–phenotype correlation. We also show, for the first time to our knowledge, mutations in MEK1 in patients with NS. Interestingly, three of our patients harbour the same D67N mutation but different phenotypes, emphasising intrinsic phenotypical variability of the mutation.
Somatic mutations in KRAS and BRAF have been identified in 7% and 15% of tumours, respectively. CS is associated with a high malignancy rate, mainly rhabdomyosarcoma, usually occurring before 6 years of age.23 Malignancies are reported in 13% of HRAS-mutated CS; risk may vary with the mutation.10 NS is associated with juvenile myelomonocytic leukaemia (JMML) in about 1–2% of cases, and possibly with an excess of childhood acute lymphoid and myeloid leukaemias. At least two patients with CFC and a BRAF mutation developed an acute lymphoblastoid leukaemia.13 24 Cancer has only been reported in two patients with CFC: one rhabdomyosarcoma in a patient with no molecular confirmation25 and hepatoblastoma in a patient with MEK1 mutation.26 Although some of our patients harbour mutations that have been reported in tumours, none has developed malignancies to date, including the patient with KRAS G12S, who is now close to 9 years old. This sporadic KRAS mutation is frequently associated with tumours and leukaemias, and has recently be reported in association with spontaneously improving JMML.27 G12S could thus induce a milder tumorigenic phenotype than other KRAS G12 mutations. Because of their young age, these children remain at a theoretical high risk of developing some malignancies. As all are sporadic cases, we cannot exclude mosaicism in these patients; however, they all display the classic phenotypic features, and the presence of the mutation was confirmed in fibroblasts in the patient harbouring G12S.
Based on current knowledge of the genotype–phenotype correlations, three clusters of genes can be classified. The first group comprises genes ouside the RAS–RAF–MEK backbone, which encompasses those upstream of RAS and those that could interact with the mainstream cascade. Most, if not all patients with PTPN11 mutations have NS or LEOPARD syndrome. Neurofibromatosis type 1 (NF1) is a neurocutaneous syndrome due to mutation in neurofibromin, a GTPase activating protein promoting RAS inactivation. When patients with NF1 have dysmorphism, they disclose a mild NS gestalt. The initial data about SOS1 seem comparable with those obtained for PTPNII, leading to the hypothesis that mutations in this group usually lead to an NS phenotype, with a low rate of mental impairment and a low rate of keratinisation disorder, but a tendency to patchy skin hyperpigmentation, and, at least for NF1 and PTPN11, a slightly increased risk of leukaemias, biased towards JMML.
The second group comprises KRAS and the cascading genes downstream. Mutations in these genes usually affect the cognitive functions, have more influence on somatic growth, skin redundancy and looseness, keratinisation (except for KRAS) and hair development, but they rarely affect pigmentation and usually result in a CFC phenotype. Malignancy risk appears to be low, but could include the commoner leukaemias rather than JMML.
The third group is restricted to HRAS. Diffuse hyperpigmentation, ulnar deviation of the wrists, papillomata, chaotic atrial fibrillation and tendency to soft-tissue tumours are the most distinguishing endophenotypes in this group.
Unravelling the molecular bases of CS, NS and CFC raises nosological problems. Do we have to base a diagnosis on clinical criteria, and accept genetic heterogeneity as a “curiosity”, or should we change to a molecular-based definition of the three entities? A molecular definition implies that a molecular diagnosis is possible (which is not the case for the 50% of patients for whom no mutation can be detected) and available (which is not the case for most patients worldwide, for practical reasons). Clinicians would have to accept that two patients with the same clinical phenotype could have two different diagnoses and that each gene-based syndrome is highly variable in its expression and shows wide overlap with the others. Obviously, a molecular-based definition can be confusing for parents, caregivers not accustomed to the subtleties of molecular dysmorphology, and even geneticists. For the NS–CFC continuum, there is to date no obvious reason to abandon clinically based diagnosis, although we probably need to redefine the border between both disorders. On the other hand, a molecular definition is appropriate when prognosis and risks for some complications (with implication for the daily care) depend upon the genotype more than on the phenotype. This is typically the case for CS, for which cancer risk and the risk for arrhythmia or vascular anomalies is clearly genotype-dependent. For that reason, we strongly recommend limiting the diagnosis of CS exclusively to patients carrying HRAS mutation. Patients with BRAF, KRAS, MEK1 or MEK2 mutations should be diagnosed as NS or CFC, whatever their phenotype. The term “severe CFC” could be used for those clinically resembling CS. Based on this, we decided to modify the diagnosis of patients with HRAS-negative CS from CS to CFC. Most parents accepted this change easily, as we could use the fact that the reclassification of their child was based on the newly acquired molecular data and was not a correction of an erroneous diagnosis. Interestingly, after the disclosure of our results, the French CS support group decided to change its name to “CS and CFC support group”.
We will progressively have to think of disorders in terms of mutation-specific complications, and not only in term of gene-specific phenotype, as illustrated by LEOPARD syndrome. Kratz et al12 showed that 8/19 patients with NS and myelodysplasia or JMML carried a single T73I substitution, a mutation that confers a much high risk of leukaemia than other alterations of PTPN11, even though the developmental anomalies are similar to those observed with other mutations.
Acknowledgments
We wish to thank the patients and their families who participated in this study and physicians who referred the patients reported in this paper: Drs MC Addor, A Afenjar, J Amiel, JM André, E Barouk, C Bellesme, D Bonneau, A Bottany, L Burglen, V Cormier-Daire, A David, S de Almeida, MA Delrue, B Doray, V Drouin-Garraud, T Edouard, B Gilbert, F Guiliano, R Hennekam, A Hovnanian, H Journel, M Kassis, M Le Merrer, S Lyonnet, S Manouvrier, G Morin, G Mortier, M Moutard, S Odent, L Pasquier, G Plessis, A Pennerah, N Phillip, P Sarda, Y Sznajer, J Vigneron, and C Vincent-Delorme. We also sincerely thank Michelle Rallet for secretary work and Céline Liger for technical assistance.
REFERENCES
Footnotes
The first three authors contributed equally to this work
Parental/guardian informed consent was obtained for publication of figures 2 and 3.
Note added in proof: Since submission of this manuscript, Gripp et al28 has reported a series of eight patients with BRAF and five with MEK1 mutations, for which the clinical diagnosis was felt to be CS. Comparison with HRAS-mutated showed similar trends to our own observations. They also favoured a molecular definition of CS.
Funding: The work was supported in part by the Programme Hospitalier de Recherche Clinique (grant PHRC-AOM02 004).
Competing interests: none declared.
- Abbreviations:
- CFC
- cardio-facio-cutaneous
- CHD
- congenital heart defects
- CS
- Costello syndrome
- GEF
- guanosine exchange factor
- JMML
- juvenile myelomonocytic leukaemia
- NF1
- neurofibromatosis type 1
- NS
- Noonan syndrome