Article Text

Abstract
Background: Mitochondrial DNA (mtDNA) mutations cause a wide range of serious genetic diseases with maternal inheritance. Because of the high transmission risk and the absence of therapy in these disorders, at-risk couples often ask for prenatal diagnosis (PND). However, because heteroplasmy load (coexistence of mutant and wild-type mtDNA) may vary among tissues and with time, the possibility that a single fetal sample may not reflect the whole neonate impedes prenatal diagnosis of mtDNA diseases.
Methods: We performed 13 prenatal diagnoses for the NARP (neurogenic weakness, ataxia, retinitis pigmentosa) m.8993T→G mtDNA mutation (p.Leu156Arg) in the ATP synthase subunit 6 gene. Analyses were performed on chorionic villous (CVS) and/or amniocyte samples carried out at various stages of pregnancy, using a method enabling quantification of low DNA amounts.
Results: Maternal mutant loads ranged from 0 to 75% in blood and had no predictive value for the fetus status, except for women with no detectable mutant DNA, whose fetuses were consistently mutation-free. In 8/13 PND, mutant load was <30%. These children are healthy at 2–7 years of age. In 5/13 PND, mutant load ranged from 65 to 100%, and parents preferred to terminate the pregnancies (15–22 weeks of gestation). Single-cell analysis of 20 trophoblastic cells and 21 amniocytes isolated from two affected fetuses found an average mutant load close to the overall CVS and amniocyte mutant load, despite striking intercellular variation. The m.8993T→G mutant loads, assessed in 7, 17, 11, and 5 different tissues from 4 terminations, respectively, were identical in all tissues from a given individual (mean (SD) 78 (1.2)%, 91 (0.7)%, 74 (2)%, and 63 (1.6)% for the 4 fetuses, respectively).
Conclusions: Our results indicate that the placental/amniotic mutant loads do reflect the NARP mutant mtDNA load in the whole fetus, even when the sample amount is small, and suggest that heteroplasmy level remains stable during pregnancy, at least after 10 weeks of gestation. Although these data establish the feasibility of PND for this mutation, assessing more precisely the correlation between mutant load and disease severity should further help in interpreting PND results.
- MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes
- NARP, neurogenic weakness, ataxia, retinitis pigmentosa
- OMIM, Online Mendelian Inheritance in Man
- PND, prenatal diagnosis
- CVS, chorionic villus
- NARP
- neurogenic weakness
- ataxia
- retinitis pigmentosa syndrome
- 8993T→G
- heteroplasmy
- mtDNA
- fetus
- prenatal diagnosis
Statistics from Altmetric.com
- MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes
- NARP, neurogenic weakness, ataxia, retinitis pigmentosa
- OMIM, Online Mendelian Inheritance in Man
- PND, prenatal diagnosis
- CVS, chorionic villus
- NARP
- neurogenic weakness
- ataxia
- retinitis pigmentosa syndrome
- 8993T→G
- heteroplasmy
- mtDNA
- fetus
- prenatal diagnosis
Oxidative phosphorylation disorders are among the most common inborn errors of metabolism, and around 20% are ascribed to maternally inherited mtDNA mutations.1 Coexistence of wild-type and mutant mtDNA in a same individual (heteroplasmy) and mtDNA mitotic segregation usually generate temporal and regional variation of the mutant load. Accumulation of mutant mtDNA molecules in affected tissues is also thought to explain the progressive nature of mtDNA disorders. The two most common mtDNA disorders are NARP (neurogenic muscle weakness, ataxia, retinitis pigmentosa; OMIM 551500) syndrome and MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes; OMIM 540000) syndrome. NARP syndrome is mainly caused by the m.8993T→G NARP mutation2 in the ATP synthase subunit 6 gene (p.Leu156Arg). Another mutation at the same codon (m.8993T→C) leads to a phenotype considered as less severe. These mutations may also result in a different phenotype, so-called Leigh syndrome (OMIM 256000), an early-onset progressive neurodegenerative disorder with characteristic neuropathological features including focal, bilaterally symmetrical spongiform lesions, especially in the thalamus and brainstem regions. Studies of patients with m.8993 mutations have shown that clinical symptoms correlate with the mutant load.3–5 All people with the 8993C mutation and NARP or Leigh syndrome have a mutant load >80%. Most people with the 8993G mutation and severe NARP or Leigh syndrome have mutant loads of ⩾75%. However, patients with this mutation and “mild” NARP phenotype (problems with night vision and proximal muscle weakness) may have mutant loads ranging from 10 to 90%.3
The recurrence risk for NARP and/or Leigh is high in offspring from women carrying the m.8993T→G mutation, and this risk increases with the maternal blood mutation load.3,6 Only few data are available to date on embryofetal analysis for NARP mutations.7,8,9,10,11 In each case, extreme mutant loads (<10% or >80%) were reported, in agreement with the “bottleneck” theory, which suggests that only a small number of mtDNA molecules “selected” during oogenesis will be the founder of an individual. However, the size of the bottleneck remains strongly debated6,12,13 and seems to depend on the type of mtDNA mutation.14,15 Moreover, because heteroplasmy varies among tissues and/or with time, the mutant load quantified in a single fetal sample may not reflect the mutant load at birth. Although NARP mutant load does not show any age-related or tissue-related variation in the postnatal period,16 the lack of data about m.8993T→G segregation during the antenatal stages has hampered the development of prenatal/preimplantation procedures. Furthermore, in a recent study, quantification of different mtDNA populations (carrying various polymorphic variants) in multiple biopsies from control placentas has indeed shown intra-tissue heterogeneity of heteroplasmy loads,17 predicting that prenatal diagnosis of mtDNA disorders, when performed on an insufficiently large chorionic villous sample (CVS), might give a false result. To investigate local, regional and temporal variation of the mutant load during embryofetal life, we collected tissues and cells from embryos or fetuses at risk for the NARP mutation. NARP mutant load was quantified using a standardised approach, from the infinitely great (various fetal tissues) to the infinitesimal (embryofetal cells).
MATERIAL AND METHODS
Patients
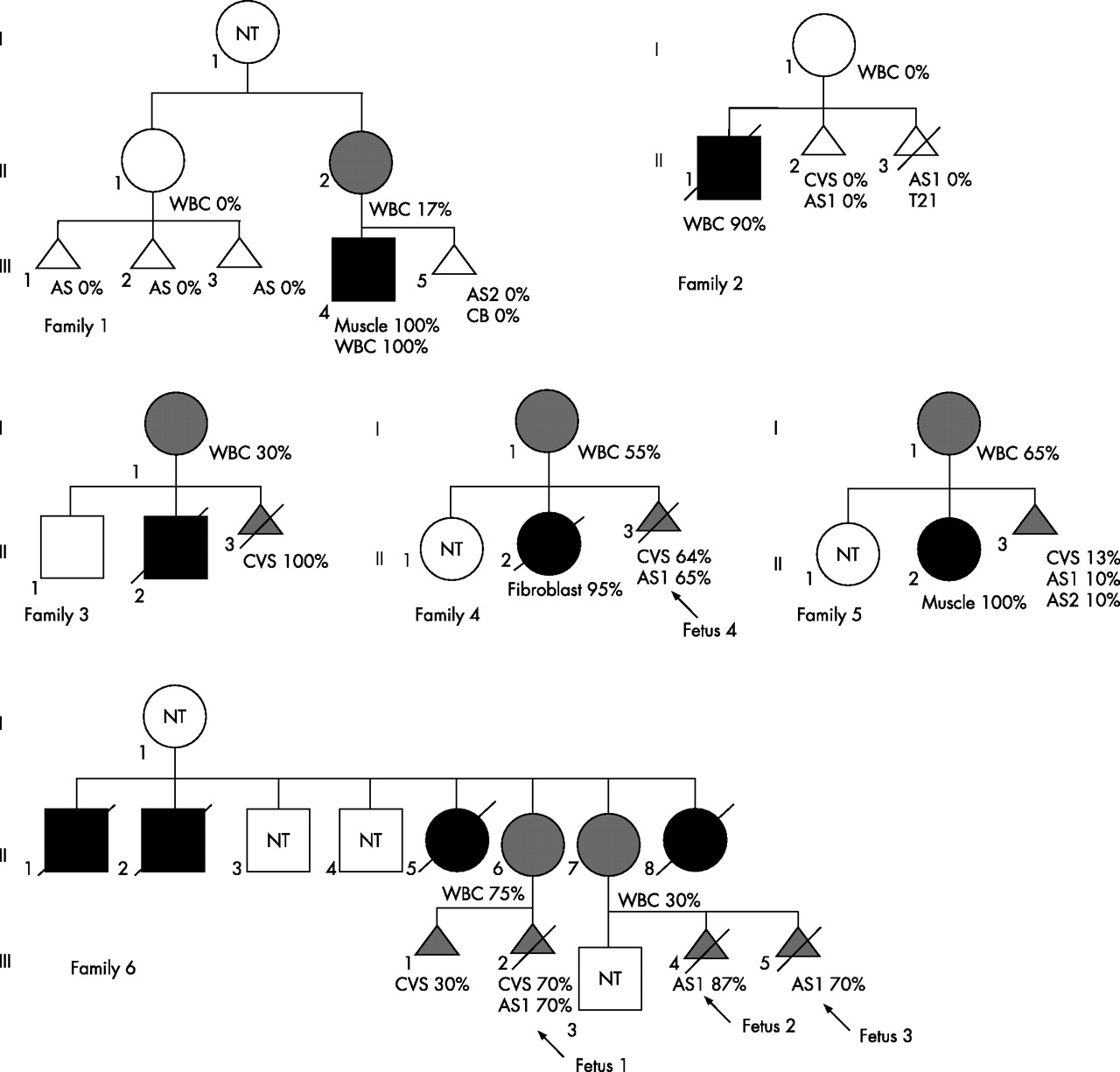
Prenatal analyses were offered to six families (fig 1) in which eight pregnant women were at risk of transmitting the NARP mutation, because they (1) had an affected child (family 1, II2; families 2–5, all I1); and/or (2) were asymptomatic carriers (family 6, II6 and II7); and/or (3) had a maternally related individual affected with NARP syndrome (family 1, II1), even though they did not carry mutant mtDNA in the blood. Informed consent was obtained from each couple after they were alerted to the uncertainties and the suboptimum nature of such analyses. Prenatal sampling was performed with the strict understanding by the family that (1) a mutant load >80% is likely to be associated with a poor outcome; (2) the test would be considered inconclusive if the mutant load was between 30% and 80%; and (3) a mutant load <30%, though being suggestive of a “good” postnatal outcome, did not guarantee an unaffected child.3,7,8,9,10,11 In the last two situations, at least one additional assessment of the fetal mutant load at a later stage of pregnancy, and when possible, from a different tissue source of fetal DNA (CVS versus amniotic fluid) was offered, in order to detect a significant variation of heteroplasmy value throughout the pregnancy.

Pedigrees of six families requesting prenatal diagnosis for the NARP m.8993T→G mutation. Black symbol, presence of symptoms ascribed to the NARP mutation; grey symbol, presence of NARP mutation without any symptom. When known, NARP mutant load are indicated under the appropriate family member. AS, amniotic fluid sample; CB, cord blood DNA; CVS, chorionic villous sample; NT, not tested; WBC, white blood cell DNA. Amniotic fluids were sampled at 14 (AS1) and 30 (AS2) weeks’ gestation, respectively.
Material
Maternal mutant load was assessed from blood and, when available, in oral mucosa cell DNA. CVS were performed at 10 weeks’ gestation. Amniocenteses were done at 14 (AS1), and 30 (AS2) weeks’ gestation, respectively. Paternal blood DNA was available in each case, to exclude maternal DNA contamination.
In four cases, material from terminations was available, enabling careful dissection of fetal tissues. Parental informed consent was obtained for fetal analyses. Small biopsies were analysed separately.
Isolation of embryofetal cells
Individual fetal cells (7 trophoblastic cells and 21 amniocytes from fetus 1, and 13 trophoblastic cells from fetus 4) were isolated and analysed separately to investigate the intercellular variation of heteroplasmy. Cells were handled in a clean laboratory with a mouth-controlled, fine heat-polished glass micropipettes, rinsed twice in two drops of phosphate-buffered saline (Sigma-Aldrich, Saint Quentin Fallavier, France) supplemented with 0.1% polyvinyl alcohol (Sigma-Aldrich) and finally deposited in 3 µl alkaline lysis buffer18 under visual control using an inverted microscope. The cells were lysed by heating for 10 minutes at 65°C.
NARP mutant load quantification
Blood and tissue DNAs were collected using a classic phenol-extraction method. NARP mutant load was quantified using a semi-quantitative fluorescent PCR restriction test, as previously described.19
In single-cell analyses, the fetal origin of the cells was assessed using two nuclear (CA)n microsatellite markers (DXS1108 and D19S112) enabling us to show (1) biparental contribution to the cell genotype, and (2) absence of maternal DNA contamination. Amplification of microsatellites markers required nested PCR. The first “outer” PCR enabled amplification of NARP mutation, combined with DXS1108,20 and D19S11221 in a multiplex PCR, using 0.5 μmol/l of each primer. Briefly, cells were amplified using 1.5 U Taq DNA polymerase (Expand Long Template), 3 μl 10× PCR buffer 2 (Roche Diagnostics, Mannheim, Germany), 0.5 μmol/l of each primer, and 2 mmol/l dNTP mix (Roche Diagnostics). PCR conditions were: denaturation at 97°C for 3 min followed by 25 (trophoblastic cells) or 27 (amniocytes) cycles of denaturation at 97°C for 20 s, annealing at 60°C for 30s and extension at 68°C for 1 min 15s, with a final extension step at 68°C for 10 min.
The first amplification product (3 µl) was subsequently transferred to 27 µl of “inner” amplification mix which contained 1.5U Expand Taq DNA polymerase, 3 μl 10× PCR buffer 2 (Roche Diagnostics), 0.5 μmol/l of each inner primer for DXS110820 and D19S112,21 and 2 mmol/l dNTP mix (Roche Diagnostics). PCR conditions were similar to those described above, except that 30 PCR cycles were performed.
RESULTS
We carried out 13 prenatal diagnoses for the NARP m.8993T→G mtDNA mutation in 8 unrelated women (table 1). Maternal mutant load ranged from 0 to 75% in blood DNA. When tested, buccal mucosa DNA loads were similar to those found in maternal blood (table 1).
Prenatal analysis of m.8993T→G
Five fetuses, originating from women with no detectable mutant DNA (I:II1, and 2:I1) were consistently mutation-free. Four children were born and were healthy at 8 months to 5 years of age, one pregnancy was terminated owing to trisomy 21.
Mothers of the eight remaining fetuses carried the m.8993T→G mutation, at least in blood. Three fetuses harboured a mtDNA mutant load <30%. In one case (fetus III5, from individual II2 in family 1), cord-blood analysis confirmed the mutation-free status (table 1). The three children are healthy at 2– 7 years of age. The mtDNA mutant load from the five remaining fetuses ranged from 65 to 100%, and parents preferred to terminate the pregnancies. The m.8993T→G mutant load was assessed in 5, 7, 11, and 17 different tissues from 4 fetuses (15–22 weeks old, table 2). The m.8993T→G mutant loads were nearly identical in all tissues from a given fetus (mean (SD) 63 (1.6)%, 78 (1.2)%, 74 (2)%, and 91 (0.7)% for the four fetuses, respectively), and close to values determined on CVS and/or amniotic fluid (65%, 70%, 70%, and 87%, respectively, table 1).
NARP mutant load in fetal tissues
Single trophoblastic cells and amniocytes were isolated from fetus 1 and 4 to investigate the intercellular variation in individual fetal samples. Genomic DNAs from trophoblastic cells (n = 20) and amniocytes (n = 21) were first shown to be fetal in origin, and free of maternal DNA, using polymorphic marker analysis. DNAs were subsequently used for heteroplasmy assessment. Substantial intercellular variation was found both for trophoblastic cells (values of 57–87% in fetus 1, and 37–84% in fetus 4) and fetus 1 amniocytes (42–95%) (fig 2). However, after pooling values from all cells analysed (for a given cell type in a given fetus), the average mutant loads (72%, 62%, and 74% for fetus 1 trophoblastic cells, fetus 4 trophoblastic cells, and fetus 1 amniocytes, respectively) were very close to the overall CVS and AS mutant load (70%, 64% and 70%, respectively).

{kind=link}
{kind=link}
Distribution of cell NARP m.8993T→G mutation levels among embryofetal cells. NARP mutant load was assessed in 7 trophoblastic and 21 amniotic cells from fetus 1 and 13 trophoblastic cells from fetus 4. White bars, tissue averages (central bar) plus (top bar) and minus (lower bar) 1 SD. Vertical bars depict maximum (top) and minimum (bottom) values for each sample.
Finally, mtDNA mutant load, assessed in six fetuses (fetus II2 from family 2, II3 from family 5, and fetuses 1–4), using at least two different samples taken at various stages of pregnancy, did not show substantial temporal variation (tables 1 and 2).
DISCUSSION
Very few data are available to date on the segregation of normal and mutant mtDNA during the prenatal period in humans. We report 13 PND, using a highly sensitive semi-fluorescent PCR test, for 8 women at risk of transmitting the m.8993T→G mutation to their offspring.
No mutant mtDNA was found in blood from two pregnant women (II1 from family 1, and I1 from family 2). However, a prenatal analysis was offered, taking into account the optional presence of mutant mtDNA in their oocytes. Failure to detect mutant mtDNA in two later fetuses from I1 in family 2 suggested a “de novo” occurrence of m.8993T→G in her first child, a common finding for this mutation.3 In these two families, four children were born and are healthy to date with a follow-up of 8 months to 5 years, and one pregnancy was terminated because of trisomy 21. These data corroborate those of a previous study on families where another mtDNA mutation, m.3243A→G, segregates. Three women with no m.3243A→G mutation in leukocytes, hair follicles and urinary tract cells were nonetheless considered to be at risk of carrying this mutation in their oocytes, owing to their position in the family pedigree. Prenatal analyses carried out on their four fetuses consistently failed to detect any mutant DNA.22 Taken together, these results suggest that, although there is still a risk of passing mtDNA mutations to children for women who do not carry the mutation in blood, it is likely to be small. However, additional data are required before drawing a firm conclusion on this point.
In our study, six pregnant women were shown to carry m.8993T→G, with similar mutant loads in blood and buccal mucosa (when available), in agreement with previous data showing no dispersion of NARP mutant load among tissues in postnatal period.3 In total, 7/8 fetuses carried the mutation, as emphasised by previous studies showing preferential transmission of mutant versus wild-type DNA for this mutation.3,6 No correlation between maternal and fetal mutant load was found in this series. This point apparently refutes previous data showing that the recurrence risk in offspring can be calculated based on the maternal mutant load.3,23 These conflicting data might in fact make sense if the mutation load in the mother is correlated with the recurrence risk in offspring in the postnatal period, although, owing to the genetic bottleneck, it will not be predictive of the mutation load in an individual fetus.
Although 4/7 fetuses had a mutant load >80% or <30%, heteroplasmy values were in the intermediate range for the remaining ones (64–70%). To our knowledge, intermediate values for m.8993T→G heteroplasmy have to date not been reported in the prenatal period. Based on the absence of data correlating intermediate mutant load in the prenatal period with the postnatal outcome, parents in our study chose to terminate the pregnancies. These fetuses questioned the size of the genetic bottleneck. The few oocytes from patients with “NARP” that were typed by some groups for NARP mutant load consistently showed markedly skewed mtDNA segregation (0% versus >90%) suggesting a tight bottleneck size.11,14 Should skewed segregation of the NARP mutation be the rule during oogenesis, intermediate mutant loads observed in the fetuses of this study might be accounted for by post-zygotic drift, possibly influenced by individual factors and/or the variant type. In agreement with this, we previously found that mtDNA molecules carrying the NARP mutation segregate differently from molecules with a homopolymeric cytosine tract polymorphism (HV2), suggesting that a particular variant might affect the mtDNA molecule segregation.11
NARP mutant load did not vary across different tissues from each of the four fetuses studied, and furthermore correlated with that detected in CVS or amniocytes, irrespective of the mutant load ranging from 57 to 94%. Similar results were found previously by other groups, after analysis of several tissues from three fetuses with very high NARP mutant loads (>85%).7,8
To date, no information is available on term-related variation of the NARP mutant load during prenatal life. We did not find any temporal variation of the NARP mutant load in six fetuses who had at least two samples taken at different stages of development. A recent report on the distribution of mtDNA polymorphisms in several CVS fragments sampled from various regions of a placenta showed that heteroplasmy may vary inside a placenta by up to 20%,17 suggesting that an insufficiently large CVS may be unrepresentative of the whole placenta. In that report,17 the authors suggested that neighbouring regions of placental tissue are arranged in “patches” or “clones”, arising from different cells in the early embryo, so that different patches would carry a different mutant load. Because such a situation might seriously affect the reliability of PND for mtDNA disorders, we quantified the NARP mutant load in 41 trophoblastic and amniotic single cells, and compared the results with those provided by DNA analysis from the overall CVS or from a fresh amniotic fluid sample. We found strong intercellular variations, suggesting that the analysed cells originated from different “patches”. However, pooling data from only a few trophoblastic cells resulted in a mean value of heteroplasmy very close to that achieved through analysis of the whole CVS. Analyses of mtDNA from amniotic cells gave similar results. It can be hypothesised that the small number of analysed cells (7 in fetus 1, and 13 in fetus 4) is in fact representative of the various “patches” constitutive of the CVS.
In conclusion, our results indicate that the placental/amniotic mutant loads do reflect NARP mutant mtDNA load in the whole fetus even when the sample amount is small, and that the heteroplasmy level remains stable during pregnancy, at least after 10 weeks of gestation. These data establish the feasibility of PND for the m.8993T→G mutation, and furthermore suggest that reliable information on the “overall” fetal mutant load can be provided by a single DNA analysis, irrespective of the tissue analysed and of the stage of pregnancy. The correlation between prenatal mutant load and disease severity remains to be assessed more precisely to help in interpreting PND results, particularly when an intermediate mutant load is found. Follow-up of our cohort of patients born after prenatal diagnosis provided a preliminary indication, even though we elected not to check for the presence of the mtDNA mutation in them, because of ethical considerations. All these children, shown to harbour <30% mutant load in the prenatal period, are healthy at 2–7 years of age. A longer-term follow-up in these children will help to draw conclusions on the predictive value of the NARP prenatal diagnosis for postnatal outcome.
Acknowledgments
J Steffann was supported by l’Association Française contre les Myopathies (grant number 10554). This work was supported by the Mitocircle contract from the European Commission (grant no 005260).
REFERENCES
Footnotes
-
Competing interests: None declared.
-
Published Online First 1 June 2007