Article Text

Abstract
Birt-Hogg-Dubé syndrome (BHD) is an autosomal dominant cancer syndrome characterised by benign skin tumours, renal tumours, and spontaneous pneumothorax. The gene has been mapped to chromosome 17p11.2 and recently identified, expressing a novel protein called folliculin. We report the clinical and genetic studies of four sporadic BHD cases and four families with a total of 23 affected subjects. Haplotype analysis of these families using BHD linked markers showed they did not share the same affected alleles, excluding common ancestry. Mutation analysis of the BHD gene identified two germline mutations on exon 11 (c.1733insC and c.1733delC) in three of four families as well as two of four sporadic cases. A novel somatic mutation, c.1732delTCinsAC, was detected in a BHD related chromophobe renal carcinoma. Our results confirmed the (C)8 tract in exon 11 as a mutational hot spot in BHD and should always be considered for future genetic testing. Our observation also indicated that the second hit (of Knudson’s two hit theory) in some BHD related tumours is in the form of somatic mutation rather than LOH. In a large French family in which eight affected subjects carry the c.1733delC mutation, a phenocopy who has multiple episodes of spontaneous pneumothorax was identified. A total of five mutation carriers (aged between 37 to 66) did not have any evidence of BHD features, suggesting either reduced penetrance or late age of onset of the disease. In addition, six out of eight affected subjects who have positive germline mutation have confirmed neoplastic colonic polyps, indicating that colorectal neoplasia is an associated feature of BHD in some families. Our studies have observed several interesting genetic features in BHD: (1) the poly (C) tract in exon 11 as a mutational hot spot; (2) the existence of phenocopy; (3) reduced penetrance or late age of onset of disease; (4) association with colorectal neoplasia in some families; and (5) somatic mutation instead of LOH as the second hit in BHD tumours.
- skin tumours
- colorectal neoplasia
- BHD hot spot mutation
- Birt-Hogg-Dubé syndrome
Statistics from Altmetric.com
Birt-Hogg-Dubé syndrome (BHD, OMIM 135150) is an autosomal dominant disease characterised by the cutaneous triad of multiple fibrofolliculomas, trichodiscomas, and acrochordons, which was first described in 1977.1 Clinically, it exhibits numerous, asymptomatic, skin coloured, dome-shaped papules over the face, neck, and upper trunk. Fibrofolliculomas and trichodiscomas are both benign tumours of the pilar apparatus. Fibrofolliculomas arise from both mesodermal and ectodermal components, whereas trichodiscomas represent fibrovascular proliferations of the hair disc.2 It is difficult to distinguish fibrofolliculomas and trichodiscomas clinically, and histologically they both consist of follicular hamartomas. BHD patients sometimes lack acrochordons,3,4 which were later defined as histological variations of the trichodiscoma/fibrofolliculoma.5 Cutaneous lesions in BHD usually appear in the third or fourth decades of life and persist indefinitely.
A wide spectrum of features has also been described in BHD along with the triad of dermatological lesions.6 These include diverse classes of renal tumours such as oncocytoma, chromophobe, papillary, and clear cell renal carcinomas (RCCs).7–9 Spontaneous pneumothorax and/or pulmonary cysts associated with BHD have also been frequently reported.8,10 In addition, colonic neoplasia was also described in earlier reports.6,10–12 However, not a single case of colonic neoplasia was found in a recent study reviewing the clinical features of 152 BHD patients from 49 families.8 The same group of researchers also concluded that there is no significant risk increment for the development of colon polyps or colon carcinomas in subjects with BHD.13
The BHD gene was mapped to chromosome 17p11.2 based on genome wide linkage studies.9,14 The BHD gene was recently identified and expresses a novel protein called folliculin but its function remains unknown.15 The identification of this gene provides an opportunity for genetic testing which will lead to a better understanding of the disease. In this study, we report clinical and genetic studies of four apparently sporadic cases and four families with 23 affected subjects from France and Sweden.
MATERIAL AND METHODS
Subjects
One three generation BHD kindred with 10 affected subjects (F518) (fig 1), one two generation kindred with five affected subjects (F598), one four generation kindred with 10 affected subjects (S001),9 and one nuclear kindred with two affected subjects (S002) were studied, along with four sporadic cases (S003, S006, F641, F676). All samples were from either France or Sweden. All studies were approved by the Ethical Committee at Le Kremlin-Bicêtre University Hospital in France, the Karolinska Hospital Ethics Committee in Sweden, and the Internal Review Board of the Van Andel Research Institute in the USA. Informed consent was obtained from all participating patients and family members.

(A) Pedigree and haplotype of family F518 with BHD. Blackened bars represent the affected haplotype. + designates subjects with the c.1733delC mutation detected and – subjects without mutation. Note the four clinically unaffected subjects (III.2, III.6, III.7, and IV.1) who are BHD haplotype and mutation carriers. III.5 is a phenocopy. Part of the affected haplotype shown in the striped bar (III.1) with the absence of mutation suggests the coincidence of possible allele sharing originating from an unaffected parent (II.1 or II.2). (B) Families F598, S001, and S002 with BHD.
Blood samples were obtained and DNA was extracted from lymphocytes using the Qiagen Maxi kit (Qiagen, California, USA). A chromophobe renal carcinoma from an affected member in family S001 was also included for DNA extraction.
Clinical studies
Evaluation was carried out at several hospitals in France as well as at the Department of Dermatology and Venereology, and the Department of Clinical Genetics in Karolinska Hospital. Each patient underwent thorough skin examination and diagnosis was confirmed histologically by biopsy. Abdominal CT scans and/or renal ultrasonography were applied to detect the presence of renal tumours. Thoracic CT scans were performed to scan for abnormalities of the lungs and colonoscopy was used to detect colorectal neoplasia.
Genotyping, haplotyping, and loss of heterozygosity (LOH) analysis
Ten microsatellite markers from chromosome 17p12-q11.2, D17S921, D17S1857, D17S740, D17S2196, D17S620, D17S805, D17S2187, D17S1871, D17S783, and D17S1824, were genotyped on all lymphocytes and tumour DNA. PCR was performed in a 7.5 μl reaction volume containing 0.17 μmol/l each of fluorescence labelled forward and unlabelled reverse primer, 10 mmol/l Tris-HCl (pH 8.3), 50 mmol/l KCl, 4 mmol/l MgCl2, 0.3 U AmpliTaq Gold polymerase (ABI), 0.25 mmol/l dNTPs (Invitrogen), and 15 ng of genomic DNA. PCR was performed using PE 9700 thermocyclers with an initial 10 cycles (15 seconds at 94°C, 15 seconds at 55°C, 30 seconds at 72°C) followed by 20 cycles (15 seconds at 89°C, 15 seconds at 55°C, 30 seconds at 72°C). PCR amplified products were then denatured and run on an ABI 377 genetic analyser (Applied Biosystem). Allele sizes were assigned using Genescan v 3.1 and Genotyper v 2.5 software (Applied Biosystem). Pedigrees were drawn using Cyrillic v 2.1 and haplotypes were confirmed with Genehunter v 2.1.16 LOH was performed by comparing the genotypes of tumour DNA with matched lymphocyte DNA. LOH was determined as previously described.17
Mutation analysis
For mutation analysis of BHD, we used the primer sequences and PCR conditions reported by Nickerson et al15 to amplify all 14 exons. Amplicons were verified by gel electrophoresis before purification with Multiscreen PCR cleanup plates (Millipore). Sequencing reactions were performed using Big Dye Terminator (Applied Biosystem), purified through Sephadex G-50 (Pharmacia), and run on an ABI 3700 genetic analyser. We aligned and analysed all sequences by Blast 2 Sequences18 as well as manually.
RESULTS
Clinical data
The clinical data of all affected subjects, except those from family S001,9 are summarised in table 1. Family F518 (fig 1A) is a large three generation family first described by Binet et al11 in 1986. This family has 10 members with typical cutaneous fibrofolliculomas and was therefore considered as affected with BHD. Multiple recurrent pneumothorax occurred in six of the affected members. III.5 (aged 47) who has many recurrent pneumothorax was initially considered as affected, but was tested negative for the mutation, therefore representing a phenocopy. Regarding the occurrence of kidney tumours, apart from two benign cysts detected in III.9 (aged 71), none of the eight examined affected patients has a renal tumour. In contrast, neoplastic colorectal polyps of different histological types (villous, tubulovillous, or tubular), mainly with a mild to moderate degree of dysplasia, were observed in six of the affected patients: II.5, III.9, IV.2, IV.4, IV.5 and IV.6. II.7 died at the age of 75 years without detection of cutaneous lesions. She and her dead sister (II.4) were both obligate gene carriers (fig 1A), and the cause of death in both cases was thought to be gastrointestinal cancer of probably colonic origin, although no pathological data could be retrieved to ascertain the sites of cancer. In addition to BHD and BHD related symptoms, tonsil squamous cell cancer was found in affected member IV.6 as well as the gastrointestinal cancer mentioned above in both obligate carriers (II.4 and II.7).
Clinical features of 32 subjects from three familial and four sporadic BHD cases
Family F598 (fig 1B) is a newly described two generation BHD family from France. Five members are affected with typical fibrofolliculomas and therefore considered to be affected with BHD. The proband (II.4) has a history of right nephrectomy at the age of 20 because of clear cell renal carcinoma, then removal of isolated bilateral parotid metastases 43 years later. The other three affected subjects showed normal renal ultrasonography. None of the patients experienced pneumothorax and we did not perform colonoscopy on the patients in this family. Other cancers detected in this family are superficial spreading malignant melanoma in the leg (II.3) and lung carcinoma in a dead obligate carrier (I.1).
Family S001 (fig 1B) has been previously described9 and has 10 members affected with BHD. One subject (V.4) had a spontaneous pneumothorax and atypical cyst in his kidney, while two other members (IV.4 and V.3) have renal cell carcinoma as well as pneumothorax. Other clinical features included sarcoma dorsi, fibroadenomatosis of the breast, and multiple lipomas.
Family S002 (fig 1B) is a newly ascertained BHD Swedish family. Multiple skin lesions were detected in the father (I.1) and daughter (II.1). Both have pneumothorax but no kidney lesion. Colonoscopy was not performed.
S003 and S006 are sporadic cases of BHD. In both cases, both of their parents have died and their sibs did not show any sign of cutaneous lesions or other BHD related features. Fibrofolliculomas were found in both patients, but renal ultrasound did not show any abnormalities. One episode of pneumothorax, unclassified colon polyps, and breast sarcoma were detected in subject S006.
Sporadic case F641 is a white male with multiple face and neck fibrofolliculomas. His father has died and he has lost contact with his mother. At the age of 35, he had a right nephrectomy where an 8 cm “malignant oncocytoma” infiltrating the capsule and locoregional nodes was dissected. He was then treated with BCG vaccine immunotherapy. In 1999, a thoracic CT scan of this patient showed a 3 cm metastasis of the upper right lobe of the lung and many bilateral emphysematous blebs. Resection of a segment of the lung showed renal tumour metastasis with papillary structures and many infrapleural emphysematous blebs. Reinterpretation of histological slides of the initial tumour gave the diagnosis of an unclassifiable malignant Fuhrman grade 4 renal cell tumour with a preponderant eosinophilic contingent. Colonoscopy was not performed in this patient.
Case F676 is a white female whose parents did not have BHD syndrome. Both her parents have died. She was diagnosed with BHD at age 51, owing to the presence of multiple fibrofolliculomas on her face and hands. This patient has neither renal tumour nor pneumothorax. Colonoscopy performed at the age of 61 showed absence of lesions.
Mutations
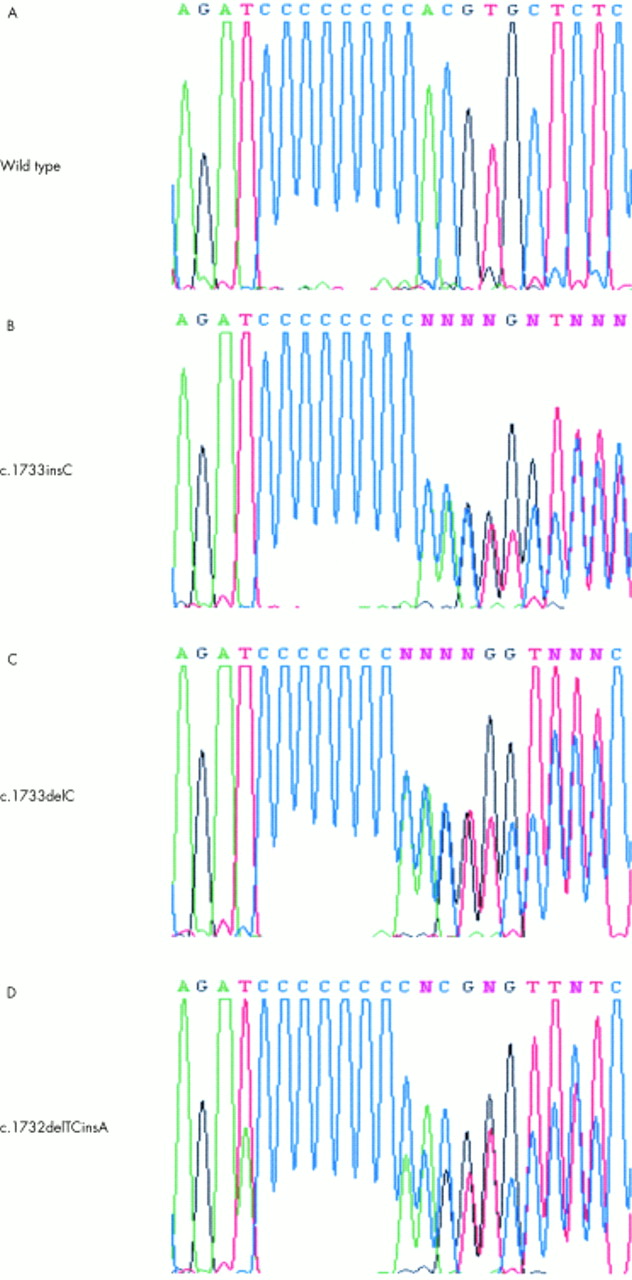
Three familial cases (F518, F598, S002) and two sporadic cases (S003, F641) showed mutations, all in exon 11. A c.1733insC (fig 2B) germline mutation was detected in S002 and S003, while all affected subjects from a family with colonic neoplasia (F518) have a c.1733delC (fig 2C) germline mutation, along with F598 and F641. The C deletion mutation in F518 also cosegregated with four BHD haplotype carriers (III.2, III.6, III.7, and IV.1, fig 1A). The same pattern of cosegregation was also observed in one BHD haplotype carrier (II.2) in family F598. A novel mutation, c.1733delTCinsA (fig 2D), in exon 11 not reported before was detected in the chromophobe tumour from an affected subject (V.3) from family S001, where no germline mutation was present in any affected members. Two isolated cases (S006 and F676) also did not show any germline mutation.
{kind=link}
{kind=link}
Representative BHD sequence of normal (A) and affected subjects (B, C), as well as a novel c.1732delTCinsA mutation (D) detected in a chromophobe tumour. These results support the poly C tract as a mutational hot spot in exon 11.
Haplotypes and LOH
All genotype and haplotype results confirmed the BHD region that was previously mapped.9,14 A detailed haplotype for family F518 is shown in fig 1A. In this family, four unaffected subjects (III.2, III.6, III.7, and IV.1) shared the same haplotype as the rest of the affected subjects and are defined as BHD haplotype carriers. The four families did not share the same affected haplotype (table 2). In family S001, no LOH was found in the chromophobe renal carcinoma of an affected member who has the disease haplotype (V.3) (data not shown).
Disease haplotype of four familial BHD cases showed that they did not share the same affected alleles. Matching mutation analysis results are also included. BHD is located between D17S740 and D17S2196. Bold numbers represent the affected haplotype
DISCUSSION
BHD is a classical hereditary cancer syndrome. The identification of the BHD gene facilitates the genetic characterisation of the patients and their tumours, including genotype-phenotype correlation, as well as functional studies of the gene. In the present study, three different frameshift mutations in exon 11, c.1733insC, c.1733delC, and c.1732delTCinsAC, were identified. The c.1733insC and c.1733delC mutations were germline mutations detected in 75% (3/4) of familial cases and 50% (2/4) of sporadic cases. Unfortunately DNA from the parents of the sporadic patients was unavailable for further analysis to determine if they were de novo mutations. The c.1732delTCinsAC mutation is a novel somatic mutation that was detected in a chromophobe renal carcinoma of a BHD patient. All mutations involve a poly C tract (nt 1733-1740) in exon 11, confirming the hypermutable (C)8 tract recently reported, where 18 c.1733insC mutations and nine c.1733delC mutations were identified in 62 BHD patient samples.15 This instability appears to be “slippage” during DNA replication, resulting in gains or losses of repeat units,21 such as in BRCA1, NF1, and APC, causing cancer predisposition.22,23,24 To date, besides exon 11, three other exons (7, 9, 12) were found to harbour BHD mutations.15 All four exons should be the focus of genetic testing, leading to saving in cost, labour, and time.
Haplotype analysis of four BHD families (table 2) showed a different disease haplotype within each family, implying that they are unlikely to share a common ancestor and ruling out the possibility of a founder effect in their mutations. All affected members in family S001 shared the same disease haplotype, which was mapped on chromosome 17p11.2,9 but none of them have germline mutation. This can be explained by (1) mutation in the promoter region; (2) a small germline deletion between marker D17S740 and D17S2196; or (3) a second BHD gene located in the vicinity of the mapped locus. The difference between family S001, which has no germline mutation, and those cases which have mutations (F518, F598, S002, S003, and F641) is the presence of breast tumours in some BHD members in S001. Interestingly, breast tumour also occurred in S006, who is a sporadic case without any germline mutation.
The majority of hereditary cancer syndrome genes follow the paradigm of a tumour suppressor gene or Knudson’s two hit theory.19 Tumours usually carry a copy of an inactivating germline mutation and lose the other copy via loss of heterozygosity (LOH), methylation, or somatic mutation. LOH is most commonly found in hereditary tumours, but in BHD only 17% of hereditary renal tumours showed LOH.20 In the only BHD related chromophobe tumour available in this study, LOH could not be detected; instead a somatic inactivating mutation, which is absent in the matched constitutional DNA, was identified. The somatic mutation represents the second hit of Knudson’s theory, and should be screened for in BHD related tumours without LOH. Other mechanisms, such as hypermethylation, may also be involved and merit further investigation.
Several groups have reported cases of colorectal neoplasia in BHD families,6,10–12 but a more recent clinical study8 showed the absence of colorectal neoplasia in 152 patients from 49 families. One study13 showed statistical non-significance comparing the presence of colon cancer in 111 BHD affected and 112 BHD unaffected subjects, as well as colon polyps of 45 BHD affected and 38 BHD unaffected subjects, concluding that there is no risk increment for the development of colon polyps or colon carcinoma in BHD patients. In the present study, we identified six family members with true neoplastic colonic polyps and two with probable colon cancer, indicating that colorectal neoplasia is involved in this particular BHD family. It is not uncommon to find certain unique clinical features associated with certain families in hereditary cancer syndromes. Environmental or additional genetic factors may be the possible cause. The latter would suggest the involvement of a “modifying” gene(s) which is related to BHD, or a distinct colorectal neoplasia related gene which happened to cosegregate with the BHD gene in some of the family members. Additional studies will be necessary fully to understand the role of BHD in colorectal tumorigenesis. In addition, our present study indicates that BHD patients may have a predisposition to other malignancies, such as melanoma, as well as gastrointestinal, lung, head and neck, and breast cancers.
Subject III.5 from family F518 had multiple episodes of spontaneous pneumothorax but without any skin lesions. The absence of germline mutation suggests that she is a phenocopy. However, c.1733delC germline mutations were detected in her two brothers, III.6 and III.7 (aged 53 and 40, respectively), although they have not yet developed any symptoms of BHD. In addition, two other subjects from F518 (III.2 and IV.1, aged 66 and 37 respectively) and another from F598 (II.2, aged 41) were also found to have germline mutations but have no clinical evidence of the disease. The findings can be explained by the late age of onset of BHD syndrome, although it is also quite possible that BHD has reduced penetrance in certain people. Further and long term studies on these families are warranted to address these issues. This study also shows the importance of mutation analysis in at risk subjects. Long term follow up and genetic counselling should be provided to gene carriers.
In summary, we have described the clinical and genetic findings in 23 affected cases from four BHD families and four sporadic cases. We confirmed a hot spot for BHD mutation, the presence of phenocopies, late age of onset or reduced penetrance of BHD, association with colorectal neoplasia, and the nature of the second hit in a BHD related tumour.
Acknowledgments
The first two authors contributed equally to this work. We would like to thank all the BHD patients and family members who participated in the studies. We also thank Dr J Amiel, Dr E Beltzer-Garelly, Dr H J Bertrand, Dr C Bizollon, Professor G Benoît, Dr J M Corréas, Dr B David, Dr W Godard, Professor J P Grünfeld, Dr J P Jacquot, Professor A Jardin, Dr G Turcat, and Dr G Allégre for their very valuable help. This study was supported by grants from the French Ligue Nationale Contre le Cancer (Comité du Cher), the Swedish Cancer Society, and the Van Andel Foundation.
REFERENCES
Linked Articles
- Correction