Article Text

Abstract
Introduction: Medullary cystic kidney disease 2 (MCKD2) and familial juvenile hyperuricaemic nephropathy (FJHN) are both autosomal dominant renal diseases characterised by juvenile onset of hyperuricaemia, gout, and progressive renal failure. Clinical features of both conditions vary in presence and severity. Often definitive diagnosis is possible only after significant pathology has occurred. Genetic linkage studies have localised genes for both conditions to overlapping regions of chromosome 16p11-p13. These clinical and genetic findings suggest that these conditions may be allelic.
Aim: To identify the gene and associated mutation(s) responsible for FJHN and MCKD2.
Methods: Two large, multigenerational families segregating FJHN were studied by genetic linkage and haplotype analyses to sublocalise the chromosome 16p FJHN gene locus. To permit refinement of the candidate interval and localisation of candidate genes, an integrated physical and genetic map of the candidate region was developed. DNA sequencing of candidate genes was performed to detect mutations in subjects affected with FJHN (three unrelated families) and MCKD2 (one family).
Results: We identified four novel uromodulin (UMOD) gene mutations that segregate with the disease phenotype in three families with FJHN and in one family with MCKD2.
Conclusion: These data provide the first direct evidence that MCKD2 and FJHN arise from mutation of the UMOD gene and are allelic disorders. UMOD is a GPI anchored glycoprotein and the most abundant protein in normal urine. We postulate that mutation of UMOD disrupts the tertiary structure of UMOD and is responsible for the clinical changes of interstitial renal disease, polyuria, and hyperuricaemia found in MCKD2 and FJHN.
- medullary cystic kidney disease 2
- familial juvenile hyperuricaemic nephropathy
- uromodulin
- chromosome 16p
- gout
Statistics from Altmetric.com
- medullary cystic kidney disease 2
- familial juvenile hyperuricaemic nephropathy
- uromodulin
- chromosome 16p
- gout
Medullary cystic kidney disease 2 (MCKD2)1 and familial juvenile hyperuricaemic nephropathy (FJHN)2 constitute a group of heritable renal diseases with a common mode of transmission (autosomal dominant) and shared features including polyuria, progressive renal failure, hyperuricaemia, and gout. Both diseases are associated with interstitial pathological changes resulting in fibrosis.3,4 While corticomedullary cysts are well documented in MCKD2, their presence in FJHN is not well documented.5 The primary clinical features of MCKD2 and FJHN vary in presence and severity, complicating the diagnosis of these conditions, particularly in milder cases.5,6
Genetic linkage studies have localised genes for both MCKD2 and FJHN to overlapping regions of chromosome 16p11-13.5–9 Because of the similarities in clinical findings and localisation of a major disease gene for both conditions to an overlapping region of chromosome 16p, it has been proposed that both diseases may arise from alteration of a common gene.5 Identification of the genetic basis of MCKD2 and FJHN will resolve the question of whether these are allelic disorders and, more importantly, permit definitive diagnostic testing. Presymptomatic diagnosis will allow early detection and treatment of the disease, which will be helpful in avoiding long term complications such as gout and hypertension. It will be of especial importance when clinically unaffected subjects are considering kidney donation to affected sibs.
MCKD2 is associated with defects in urinary concentration,5 interstitial renal damage,3 and amorphous interstitial renal deposits,10 findings that could all be related to functional defects in a protein synthesised and secreted by the renal tubules. Similarly, FJHN is associated with increased tubular uric acid reabsorption11 and tubulointerstitial damage4 that could also be caused by an abnormal protein synthesised and secreted by the renal tubules. The Tamm-Horsfall glycoprotein,12 also referred to as uromodulin,13 is a glycosylphosphatidylinositol (GPI) anchored glycoprotein and the most abundant protein in normal urine. Hoyer et al14 localised expression of uromodulin to the thick ascending limb of the loop of Henle. In addition, these authors identified amorphous deposits of uromodulin in the renal interstitium of patients with medullary cystic kidney disease.10,15
Despite significant speculation, the function of uromodulin remains unclear. It has been hypothesised to form a gel which helps maintain the integrity of the loop of Henle and the countercurrent mechanism,14 to perform immunological functions related to its ability to bind to tumour necrosis factor,16 interleukin 1α, and other cytokines,17 and to prevent urinary tract infections owing to its ability to prevent Escherichia coli from binding to human kidney cells.18 Uromodulin is also reported to have an antioxidant effect19 and to inhibit calcium oxalate crystallisation.20 In canines, uromodulin is reported to function as a novel carrier for vitamin A in the urine.21 Since it was first isolated from the urine via a salt precipitation technique by Tamm and Horsfall in 1950,12 there has been abundant speculation regarding the physiological function of this protein, and its role in renal disease has remained undetermined.
Here we report genetic linkage and mutational analyses of members of three families segregating FJHN and one family segregating MCKD2. As a result of linkage studies of two of these families, we were able to refine the candidate region for the FJHN gene locus to a region of ∼1.7-Mb. Using bioinformatic methods, we developed a physical map of the candidate region, permitting identification and localisation of six genes. Direct sequence analysis of candidate genes within this interval resulted in identification of four different mutations in exon 4 of the uromodulin (UMOD) gene in affected members in three families with FJHN and one family with MCKD2. Family members carrying the mutation suffered from hyperuricaemia, decreased fractional excretion of uric acid, enuresis, and chronic renal failure. These findings confirm that FJHN and MCKD2 result from allelic mutations of a common gene. Additionally, these results confirm speculation that aberration of uromodulin, the most common protein in urine, results in decreased ability to concentrate the urine with resulting hyperuricaemia, and chronic progressive interstitial renal disease.
PATIENTS AND METHODS
Pedigrees and diagnosis
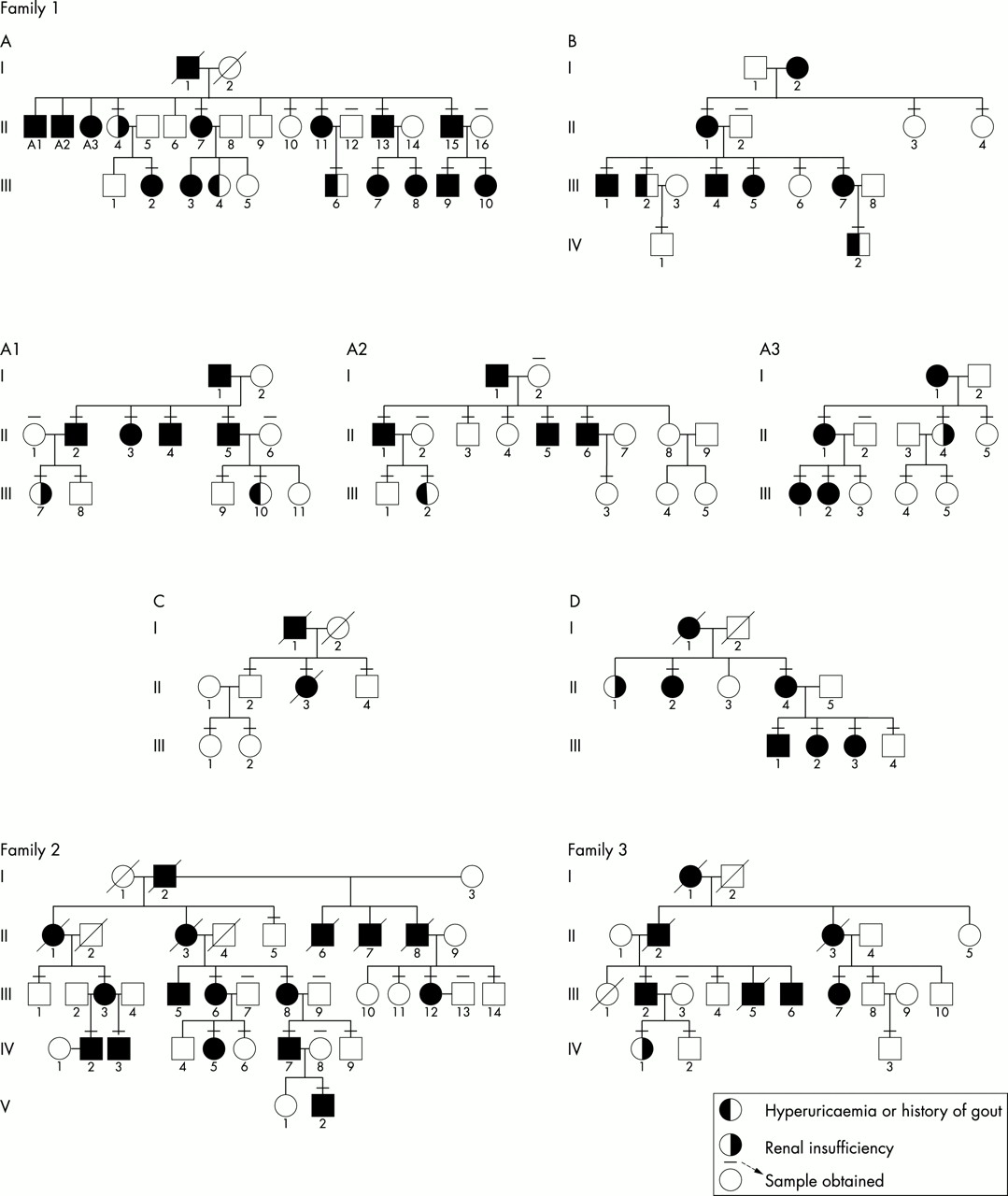
Study participants were obtained from four families. Family 1 was a large multigenerational family in which the disorder was traced back seven generations. The family tree contains more than 300 members and was too large for the entire pedigree to be depicted. Fig 1 shows the pedigree for selected portions of the family in whom the majority of samples were obtained. This family had a long history of hyperuricaemia, reduced fractional excretion of uric acid, and renal failure, inherited in an autosomal dominant fashion, with clinical findings consistent with FJHN. Family 2 was a large multigenerational family that also segregated FJHN as a highly penetrant autosomal dominant trait. Family 3 has previously been reported to suffer from medullary cystic disease, hyperuricaemia, and gout,3 inherited in an autosomal dominant fashion (fig 1). A sample was obtained from one affected family member from family 4. Family 4 was previously extensively reported as suffering from familial hyperuricaemia and renal disease but no medullary cysts, findings consistent with a diagnosis of FJHN.4

Pedigrees of families studied. Family 1: more than 300 subjects have been genealogically identified over seven generations. The kindred is too large to include in total; we have indicated the nuclear families studied for this report. These families are from different parts of this extended kindred, and are indicated as subfamilies A, B, C, and D. In addition eight singletons were studied. Clinical findings in affected family members are consistent with a clinical diagnosis of FJHN in family 1. Family 2: clinical findings in this family are consistent with a clinical diagnosis of FJHN. Family 3: clinical findings and renal biopsy/necropsy reports are consistent with a clinical diagnosis of MCKD2.3
All participating family members provided informed consent to protocols approved by institutional review boards at Wake Forest School of Medicine and the University of Pittsburgh. Serum uric acid and serum creatinine measurements were performed, and 24 hour urine collections for uric acid and creatinine were obtained. The creatinine measurements were performed by the Jaffe alkaline picrate kinetic method.22 The uric acid measurements were performed on the ADVIA 1650 Chemistry System. The uric acid determination method is based on the Fossati enzymatic reaction using uricase with a Trinder-like endpoint.23 Estimates of creatinine clearance, as determined by the Cockroft-Gault formula,24 were made using the patient’s weight or ideal body weight, whichever was less. Renal insufficiency was defined as an estimated creatinine clearance less than 80 ml/min. Enuresis was defined as persistent bed wetting after the age of 4 years.
Patients were considered to be definitely affected if they met the following criteria: (1) hyperuricaemia, defined as serum uric acid levels greater than 2 SD above the age and gender adjusted norms for the population25,26 or a history of gout and current treatment with allopurinol, and (2) reduced fractional excretion of uric acid (<5% for men and <6% for women) or a reduced creatinine clearance of less than 80 ml/min. (In general, people with a creatinine clearance less than 80 ml/min will start developing a raised fractional excretion of uric acid27 and, as such, family members with renal insufficiency could not have their fractional excretion of uric acid used as a determinant of FJHN.) Family members were defined as clinically unaffected if the serum uric acid level was within 1 SD of the age and gender adjusted norms for the population.25,26
DNA marker analysis
Genomic DNA was extracted from peripheral blood by standard methods using the QIAamp blood kit (Qiagen). We performed genetic linkage studies for 90 subjects from two extended multigenerational families diagnosed with FJHN (families 1 and 2, fig 1). Available family members were genotyped for STRP (short tandem repeat polymorphism) type genetic markers spanning the candidate interval. In addition to nine previously reported STRP loci, nine novel STRP loci were developed from a 5.6 Mb physical map of the interval (figs 2 and 3) (MCG and TCH, unpublished data, available at www.sdmgenetics.pitt.edu). These marker loci were PCR amplified by use of fluorescence labelled primers, permitting genotyping by conventional methods.28 PCR products were detected by an ABI 377 fluorescent sequencer and were analysed by GENESCAN 2.1 (Applied Biosystems).

Integrated physical and genetic map of the FJHN/MCKD2 candidate region on chromosome 16p. Genetic STRP markers and their relative locations are indicated on the left. Locations of significant linkage results (lod scores >3.0) are indicated for two families in the current report (families 1 and 2) and for five other studies.1–5 Nine known STRPs and nine novel STRPs were identified, localised, and genotyped. Two novel STRP loci were identified in BAC2349B8; the position of these loci (2349B8(16)-2 and 2349B8(16)-1) are separated by 54 000 bp and their order is given in fig 3. Genetic loci identified in the region are indicated to the right of the figure.

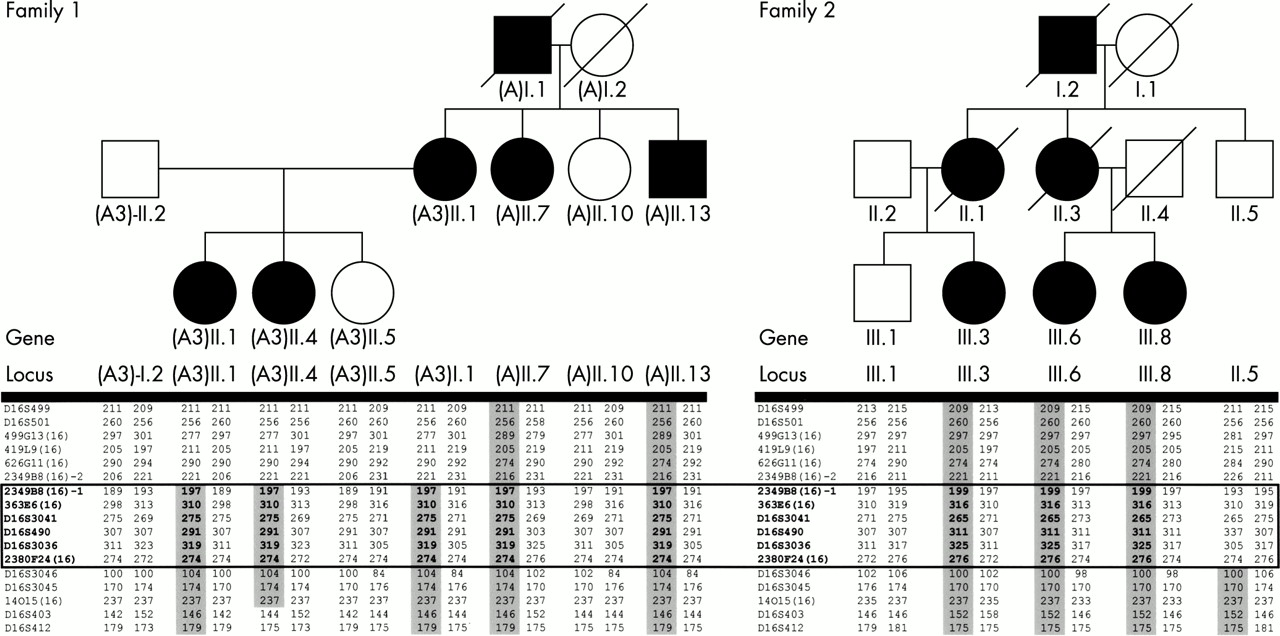
Haplotype results indicating the minimal genetic interval on chromosome 16 segregating with the FJHN phenotype in families 1 and 2. FJHN affected subjects are indicated by shaded symbols. Genetic STRP loci genotyped are listed in positional order in the left column for each family. Haplotypes segregating with the disease locus are shaded. II.5 from family 2 is unaffected, but has inherited the disease associated haplotype for the interval D16S412-D16S3046, indicating that this region does not contain the FJHN disease locus. The boxed region indicates the minimal haplotype region segregating with the FJHN in both families, indicating that the FJHN gene is within the interval flanked by 2349B8(16)-2 and D16S3046.
Parametric linkage calculations: lod scores and haplotype analysis
Sublocalisation of the candidate interval was achieved by means of genetic linkage studies and determination of the minimal region of overlap of haplotypes segregating with the FJHN trait in families 1 and 2. Standard two point and multipoint linkage analyses were performed using the VITESSE program.29 Assumptions of the linkage analyses included autosomal dominant transmission, penetrance values of 95-100%, a disease allele frequency of 0.0001, and a phenocopy rate of 1%. To permit identification of haplotypes, a physical map of the FJHN candidate gene region was developed. This map permitted precise localisation of known STRP markers within the region and allowed identification of novel STRP markers at desired locations spanning the interval.
Development of a physical map of the candidate FJGN candidate interval: STRP and gene identification
In order to identify novel STRP type markers spanning the candidate interval and to permit identification of all known and hypothetical genes within the interval, the development of a detailed physical/genetic map was initiated.30–32 The final alignment contained 67 BACs that span a 5.6 million base region. This region contains two gaps across which a BAC sequence did not align. This contig was screened for all known genes, and STRP loci were identified through the NCBI Human Genome Sequencing Site and GENEMAP 9933,34 gene and STRP loci confirmed on the BAC contig were positioned on the new map. New STRP markers were identified using the tandem repeats finder.35,36 Candidate STRP sites were then selected and primers designed using Oligo 4.2 software (table 1).
Primer sets used in the amplification of STRP loci genotyped in the FJHN/MCKD2 candidate region
Several sources of information were used to identify genes in the candidate region: the Human Genome Project Working Draft at UCSC,31 the Sanger Centre’s ENSEMBLE database,37 and Locus Link.35 NCBI BLAST 32 and ePCR were also used on the BAC contig sequence with the BLAST non-redundant and dbEST databases screened. A cDNA contig was made for each candidate gene using all information that was available at the time. The inclusion of all EST data provided for a more accurate representation of the gene. Intron/exon boundaries were determined manually using the consensus splice sequences indicated at GENIO/splice.38 Primers for amplifying candidate genes from genomic DNA were designed using data obtained from the primary contig as well as from available NCBI data (accession numbers in electronic references, NCBI Locus Link, NCBI Entrez).39,40 By means of linkage and haplotype analyses, we refined the FJHN candidate region to a ∼1.7 Mb interval. We localised five known genes to this interval. Additionally, using an integrated bioinformatic and bench lab approach, we identified one previously uncharacterised genetic locus within the interval. All exons and intron-exon boundaries of four of these genes were analysed by sequence analysis of genomic DNA from affected and unaffected family members from families 1 and 2.
UMOD exon sequencing
The genomic structure of the UMOD gene was determined bioinformatically and was confirmed by sequence analysis. Oligonucleotide primers to amplify 11 of the 12 exons, including intron-exon boundaries (table 2), were designed with Oligo 4.02 (National Biosciences). PCR amplification of the UMOD gene was performed as indicated in table 2. Amplified DNA was purified with the QIAquick PCR Purification Kit (Qiagen) and was sequenced using the BigDye Terminator Cycle Sequencing Kit on an ABI 3700 DNA Analyzer (Applied Biosystems) by the Genomics and Proteomics Core Laboratories of the University of Pittsburgh. Sequence analysis was performed with Sequencher 4.1 software (GeneCodes).
Primer sets for exonic amplification of the human UMOD gene
RESULTS
Clinical findings
Over a five year period, clinical testing was performed on 72 members of family 1. Thirty-one met strict criteria to be considered affected (hyperuricaemia with reduced fractional excretion of uric acid or renal insufficiency), 22 were diagnosed as normal, and there were 10 unaffected spouses. For nine family members, a certain diagnosis could not be made. Thirty-four subjects suffered from hyperuricaemia and 28 suffered from renal insufficiency. The pedigrees for families 2 and 3 identify all subjects who suffered from hyperuricaemia or renal insufficiency.
Renal biopsies
Pathological samples were obtained by kidney biopsy in three members of family 1. All three biopsies showed histological changes of tubular atrophy and interstitial fibrosis. Global glomerulosclerosis was present, and there was no evidence of glomerulonephritis. In family 2, a biopsy specimen of an affected female aged 39 years showed widespread tubular atrophy. Necropsy specimens have been reported for family 3.3 The first was that of a 34 year old man reportedly with tubules ensheathed by a dense acellular hyaline material.3 Medullary cysts were present. In another family member, necropsy studies again showed sheathing of the tubules by fibrous tissue. In case 3, tubules were ensheathed by dense acellular hyaline material.3 In family 4, biopsy samples showed focal tubular atrophy with interstitial fibrosis and lymphocytic infiltration. In summary, all biopsy specimens showed focal tubular atrophy with interstitial fibrosis. Necropsy reports indicated tubules ensheathed by dense acellular hyaline material. Zager et al10 and Resnick et al15 have also identified these interstitial deposits of PAS positive material in medullary cystic kidney disease. Immunostaining of these deposits was found to be markedly positive with antibody to Tamm-Horsfall protein by these authors.
Physical map of the candidate interval
Existing genetic and physical maps of the FJHN/MCKD2 candidate interval were generally poorly integrated and identified relatively few polymorphic genetic markers (STRPs) spanning the interval. This was problematical for our studies as a key marker (D16S3056) was uninformative in our families. The development of an integrated physical and genetic map of the FJHN/MCKD2 candidate interval (summarised in fig 2) permitted us to orient precisely results of previous linkage studies, to localise precisely known genes to the candidate interval, and to develop novel STRP loci. The availability of novel STRP markers permitted refinement of the candidate interval by haplotype analysis. The location of eight known and eight novel STRPs are shown in fig 2. Oligonucleotide primers and conditions used to amplify these STRPs are shown in table 1. The consensus candidate interval for most reports, including our linkage data, supported a candidate interval located in 16p13.11 (D16S499) to 16p12.2 (D16S403). It is apparent from fig 2 that while all linkage intervals reported for FJHN and MCKD2 map to chromosome 16p, not all overlap.
Linkage analyses
Results of genetic linkage analyses localised the gene for FJHN in two of our families (families 1 and 2) to an overlapping interval of ∼1.7 Mb (fig 2). For family 1, the gene was localised to an interval of ∼3.8 Mb delineated by 2349B8(16) to D16S403 (Zmax=12.5 at D16S3041, 𝛉=0.01) and for family 2 the linkage interval was ∼17 Mb between D16S404 and D16S3046 (Zmax=3.2 at D16S3041, 𝛉=0.00); D16S404 extends ∼14 Mb telomeric to D16S499. These findings were consistent with four5,6,7,9 of the previous five reports of linkage for FJHN to chromosome 16p. Our candidate interval did not overlap that of Kamatani et al,8 possibly reflecting genetic heterogeneity (they are the only group to study Japanese FJHN families).
Candidate gene evaluation: mutation analyses
Integration of all known linkage reports for FJHN with our linkage data identified an interval of minimal overlap (<0.3 Mb) from 2349B8(16) to D16S3036 for our linkage results with those of Dahan et al5 (fig 2). Our gene identification approach identified one known gene (B/K protein, NM_016524)40 and one hypothetical gene (G104, XM_091332)40 in this common interval. Direct sequence analysis of genomic DNA from affected and unaffected family members from families 1 and 2 for coding regions (including intron-exon junctions) of the B/K gene and the hypothetical gene G104 did not identify any alterations of DNA that would account for the FJHN trait in either family (data not shown).
Recognising that the definitive diagnosis of FJHN can be problematical (particularly in milder cases and in younger people), and incorrect diagnosis of family members may directly affect the boundaries of the candidate gene region, we decided to continue our analysis using only linkage and genotype data from families 1 and 2. For this reason, we excluded from our analysis subjects who could not be diagnosed as affected based on our stated diagnostic criteria. Similarly, subjects who did not have both normal renal function (calculated creatinine clearance greater than 100 ml/min) and a serum uric acid level within 1 SD of the mean adjusted for age and gender25,26 were excluded from the analysis to refine the candidate interval.
Haplotype analysis permitted us to identify the smallest common haplotype segregating with the FJHN trait in families 1 and 2 (fig 3). Our sequence analysis had excluded the known (B/K protein) and hypothetical (G104) genes from the interval 2349B8(16)-D16S3036, permitting us to refine the candidate interval to ∼1.2 Mb, from D16S3036-D16S3046. This revised candidate interval contained four genes: butyrl coenzyme A synthetase 1 (BUCS1), glycoprotein 2 (GP2), G protein coupled receptor, family C group 5, member B (GPRC5B), and uromodulin (UMOD). Sequence analyses of GPRC5B and UMOD were performed for genomic DNA from affected and unaffected family members. No coding region polymorphisms were detected in the GPRC5B sequence data (data not shown). To determine the genomic organisation of the entire UMOD gene, all available UMOD mRNA and EST data were aligned to identify any possible splice variants. Using bioinformatic approaches, we determined the genomic structure of the UMOD gene (fig 4). This approach led to the identification of 12 exons, one exon more than previously reported.41 The novel exon identified by our approach and supported by EST data is exon 2. Exons 1 and 2 are non-coding with the ATG start site in exon 3. Based upon EST data, there appear to be alternate 5′ transcription start sites so that transcription either begins with exon 1 and proceeds to exon 3 or transcription begins in exon 2 and proceeds to exon 3. In either case, the resultant protein is identical.
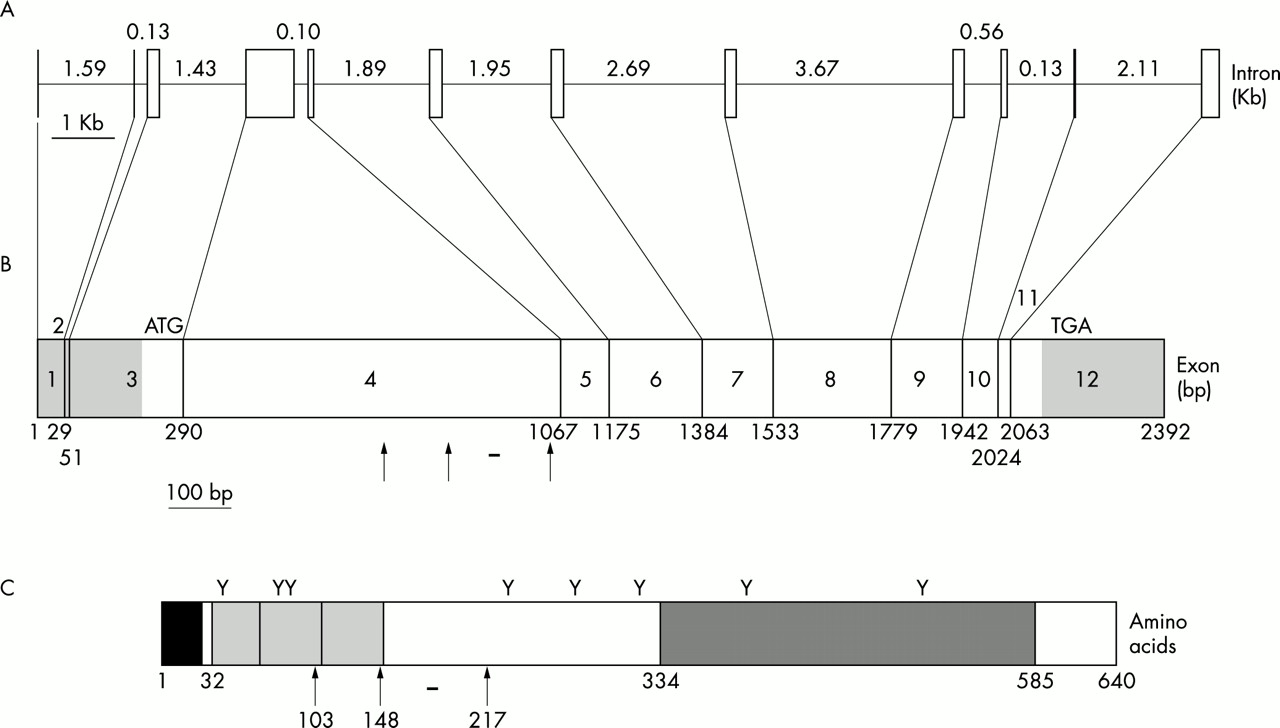
Structure of the human UMOD gene. (A) Genomic organisation of the UMOD gene. The exons and introns are represented as vertical boxes and horizontal lines respectively. The sizes of each intron are given in Rb. (B) cDNA structure of the UMOD gene. The translation start and stop codon are labelled as ATG and TGA, respectively. The 5′ and 3′ untranslated regions are shaded grey. The arrows indicate the missense mutations identified in this study. The horizontal bar indicates the deletion identified in this study. (C) Structure of the wild type uromodulin protein. The initiation met is amino acid 1. The signal peptide is shown as a black box. The EGF-like domains are shown as syntenic light grey boxes. The ZP domain is shown as a dark grey box. The eight potential glycosylation sites are shown as Y. The missense mutations identified in this study are shown as arrows with the corresponding amino acid listed below. The 9 amino acid deletion is shown as a horizontal bar.
UMOD sequence analysis was undertaken on families 1 and 2. Results of sequence analysis showed two different mutations in exon 4 of UMOD in families 1 and 2 (fig 5A, B). Mutations are described according to nomenclature guidelines.42,43 In each family (g1966_1992del in family 1 and g.1880G>A in family 2), the UMOD exon 4 gene mutation segregated completely with the disease phenotype. To evaluate the possible involvement of UMOD mutations in MCKD2, we conducted sequence analysis on three affected and five unaffected family members from a smaller family segregating MCKD2 (family 3, fig 1). Analysis of this family identified a third novel mutation (g.1744G>T) in UMOD, also in exon 4 (fig 5C). To evaluate the generality of UMOD mutations in FJHN, we performed mutational analyses on an affected member from an extended kindred previously reported.4 Analysis showed a fourth novel mutation (g.2086T>C) in exon 4 of UMOD (fig 5D).

Mutations in UMOD. The top sequence in each panel shows wild type sequence. The bottom sequence is from an affected subject. Descriptions of each mutation are given for [genomic; cDNA ; protein] in accordance with nomenclature guidelines. (A) Affected subjects in family 1 were heterozygous for a 27 bp deletion that results in the in frame deletion of amino acids 177-185 (g.1966_1992del; c529_555del; p.H177_R185del). (B) Affected subjects in family 2 were heterozygous for a missense mutation that changes a conserved cys to tyr (g.1880G>A; c.443G>A; p.C148Y). (C) Affected subjects in family 3 were heterozygous for a missense mutation that changes a gly to a cys (g.1744G>T; c.307G>T; p.G103C). (D) Affected subjects in family 4 were heterozygous for a missense mutation that changes a conserved cys to arg (g.2086T>C; c.649T>C; p.C217R).
The specific UMOD gene mutations in families 1, 2, and 3 each segregated in affected family members (FJHN in families 1 and 2, and MCDK2 in family 3). None of these mutations was identified in any of the 100 control chromosomes tested. Sequence analysis of the UMOD gene in 50 white controls (100 chromosomes) did show the presence of two silent polymorphisms within UMOD exon 4. A previously reported synonymous SNP44 located at C174 has a T allele frequency of 82% and a C allele frequency of 18% for our samples. A novel synonymous SNP located at V287 has a G allele frequency of 87% and an A allele frequency of 13%. No polymorphisms affecting the translation of uromodulin were detected in any of the 100 control chromosomes examined.
Genotype-phenotype correlation
In family 1, 36 family members carried the mutation and 26 family members did not. Thirty-two of 36 (89%) genetically affected subjects suffered from hyperuricaemia (as defined in Methods). Twenty-eight of 32 (88%) genetically affected family members had an estimated creatinine clearance less than 90 ml/min when measured after the age of 18 years. Ten of 36 (28%) subjects carrying the UMOD mutation suffered from enuresis. The fractional excretion of uric acid was less than 6% in all genetically affected men and less than 5% in all genetically affected women with an estimated creatinine clearance greater than 70 ml/min. (The fractional excretion of uric acid increases in patients as renal function declines.27) Thirty-two of 36 subjects carrying the UMOD mutation met the strict clinical criteria required to be diagnosed as affected. The remaining four were women who had normal serum uric acid levels despite low fractional excretions of uric acid. Two of these women had mild renal insufficiency. The serum uric acid levels remained normal or borderline on testing over several years in three of these women. Five family members who did not carry the UMOD mutation had serum uric acid levels which were raised but which were not greater than 2 SD above the mean.
In family 2, nine of nine patients with the mutation suffered from hyperuricaemia, and nine of nine patients suffered from renal insufficiency. In family 3, two of three family members carrying the mutation suffered from hyperuricaemia, and all three affected family members suffered from renal insufficiency.
DISCUSSION
The findings of this study establish that mutation of the uromodulin gene is responsible for FJHN and for MCKD2. Uromodulin is polymeric in its native form, composed of monomeric subunits of approximately 85 kDa, with 30% of the molecular weight due to carbohydrates and the remaining 70% due to the polypeptide chain.45 Electron microscopy showed that the high molecular weight aggregate is composed of thin, intertwining fibres with a zigzag or helical structure. Recent analysis indicates that the filaments consist of two protofilaments wound around each other, forming a right handed helix.46 Uromodulin contains a zona pelucida (ZP) domain (fig 4C), a domain which has been shown to be responsible for polymerisation of ZP containing proteins into filaments.46 Uromodulin contains a high number of cysteine residues (48 per monomer), allowing for the potential formation of 24 intramolecular disulphide bonds. These cysteine residues are very highly conserved across species (fig 6). It is interesting to note that all three of the missense mutations identified involve cysteine residues, either by substitution of another residue for cysteine (g.1880G>A: p.C148Y, g.2086T>C: p.C217R) or vice versa (g.17744G>T: p.G103C). Such substitutions may lead to important structural changes through intra- and possibly intermolecular disulphide bond formation, changes which have been documented for other proteins.47 The mutation found in family 1, predicted to result in the loss of nine amino acids (g.1966_1992del, p.H177-R185del), is also postulated to cause structural changes in the uromodulin protein. In frame deletions are known to be responsible for a number of diseases including MEN1 (multiple endocrine neoplasia type 1),48 Marfan syndrome,49 osteogenesis imperfecta,50 and pachyonychia congenita.51 Impaired protein folding can result in premature degradation, and may be a major disease mechanism in genetic deficiencies owing to missense mutations and short in frame deletions/insertions.52 It is interesting that five of the nine amino acids deleted in family 1 are highly conserved across species (fig 6). The function of this conserved region is not known, but deletion of an adjacent region of the UMOD protein is associated with decreased ability to inhibit binding of immunoglobulin light chains, resulting in cast nephropathy.53

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Alignment of the amino acid sequence of human uromodulin (GenBank accession No M17778) with the uromodulin of bovine (GenBank accession No S75958), murine (GenBank accession No NM_009470), and rat (GenBank accession No M63510). All 48 C residues are conserved and shown in bold with an asterisk. The arrows indicate the position of the missense mutations identified in this study. The nine amino acids deleted in family 1 are indicated in bold and underlined.
The present investigation provides insight into both the function of uromodulin and the nature of MCKD2 and FJHN. Owing to the similar clinical findings and linkage localisations in MCKD2 and FJHN, it has been suggested that they may represent a common disease.5 The current findings establish that FJHN and MCKD2 are allelic disorders.
The presence of hyperuricaemia in patients with FJHN has been speculated to be the result of abnormalities in the proximal tubular transport of uric acid.11 The present study contradicts this theory, as uromodulin is produced in the thick ascending limb of the loop of Henle, well after proximal tubular transport of uric acid occurs. It is likely that uromodulin is important in maintaining the countercurrent gradient and the medullary loop impermeability. The high rate of enuresis seen in the three families studied (30%) suggests that difficulties in urinary concentration are present. This decreased concentrating ability probably results in increased urinary salt and water excretion, resulting in increased proximal tubular reabsorption of uric acid. Previous studies have documented increased proximal tubular uric acid uptake in patients taking loop diuretics,54 and it is likely that a similar mechanism is responsible for hyperuricaemia in this group of patients.
Patients with this deficiency in uromodulin production also suffer from the development of chronic renal failure over time. Previous studies have well documented the abnormal deposition of uromodulin in the interstitium in patients with medullary cystic kidney disease.10 It had been speculated that this deposition was secondary to pathological changes in the medulla of the kidney,10 but it appears that these depositions are a primary result of abnormal uromodulin production. It is possible that altered uromodulin binding to various cytokines may result in fibrogenesis. Interstitial nephritis has previously been proposed as a mechanism of pathogenesis in medullary cystic kidney disease.55
In summary, mutations in UMOD are responsible for FJHN and at least some cases of MCKD2. Pirulli et al44 recently excluded UMOD as a candidate gene for a large Italian family segregating MCKD2. Although they sequenced the entire coding region, this was performed with different primer sets from those used in the current report. Methodological differences in sequencing of exon 4 may account for the different results; however, other possibilities must be considered. Deletion of an entire exon could result in PCR amplification of only the wild type allele, masking the presence of a mutation. Pirulli et al44 did not analyse the non-coding exons 1 and 2, nor the 5′ regulatory region of UMOD. It is possible that mutations in exon 1, exon 2, or the regulatory region could result in loss of uromodulin production.56,57 Alternatively, genetic heterogeneity may exist with another kidney specific gene located in the candidate interval.
Identifying the uromodulin mutation in families with FJHN and MCKD2 is helpful in furthering the clinical characterisation of these disorders. The cardinal feature of both disorders is chronic, progressive renal failure that is tubulointerstitial in origin. Hyperuricaemia was found in the vast majority, but not all family members identified with this disorder. Four women carrying the mutation had normal serum uric acid levels on repeated testing. Despite this, renal insufficiency had developed in two of these women. Similarly, medullary cysts are frequently not seen in radiological examinations of these patients. Given that hyperuricaemia is not always present in FJHN and given that medullary cysts are not always present in MCKD2, and given that these two conditions result from mutations of the same gene, we believe it is appropriate to designate these two conditions as uromodulin associated kidney disease. These findings provide exciting new insights into the function of the most abundant urinary protein and should act as a further catalyst to research on the functions of uromodulin.
Acknowledgments
The authors wish to thank the following people for their contributions: Debra McBride, Betsy McAllister, Scott Satko, Elizabeth Deterding, Mark Hinsdale, M J Pettenati, J Charles Jennette (for providing biopsy specimens), and Elizabeth Phillips. The authors wish to thank Dr Bryson Fleming and members of Asheville Kidney Associates in Asheville, NC for referral of members of family 1, and Dr Scott Stromatt for referral of the proband in family 2. This investigation was supported in part by United States Public Health Service research grant DK62252 from the National Institute of Diabetes & Digestive & Kidney Diseases and by a grant from the National Kidney Foundation of North Carolina Inc.