Article Text

Abstract
Generalised lipodystrophy of the Berardinelli-Seip type (BSCL) is a rare autosomal recessive human disorder with severe adverse metabolic consequences. A gene on chromosome 9 (BSCL1) has recently been identified, predominantly in African-American families. More recently, mutations in a previously undescribed gene of unknown function (BSCL2) on chromosome 11, termed seipin, have been found to be responsible for this disorder in a number of European and Middle Eastern families. We have studied the genotype/phenotype relationships in 70 affected subjects from 44 apparently unrelated pedigrees of diverse ethnic origin. In all subjects, hepatic dysfunction, hyperlipidaemia, diabetes mellitus, and hypertrophic cardiomyopathy were significant contributors to morbidity with no clear differences in their prevalence between subjects with BSCL1 or BSCL2 and those with evidence against cosegregation with either chromosome 9 or 11 (designated BSCLX). BSCL2 appears to be a more severe disorder than BSCL1 with a higher incidence of premature death and a lower prevalence of partial and/or delayed onset of lipodystrophy. Notably, subjects with BSCL2 had a significantly higher prevalence of intellectual impairment than those with BSCL1 or BSCLX (p<0.0001, OR 17.0, CI 3.6 to 79.0). The higher prevalence of intellectual impairment and the increased risk of premature death in BSCL2 compared to BSCL1 emphasise the importance of molecular diagnosis of this syndrome and have clear implications for genetic counselling.
- genotype-phenotype correlation
- lipodystrophy
- mental retardation
- seipin
Statistics from Altmetric.com
Congenital lipodystrophy of the Berardinelli-Seip type (BSCL, OMIM 269700) is a rare condition characterised by a near total absence of adipose tissue.1–3 Because of the absence of functional adipocytes, dietary and endogenously synthesised lipid is stored aberrantly in metabolically important tissues such as muscle and liver. This leads to severe insulin resistance and, ultimately, diabetes mellitus, which is often difficult to control. Other metabolic consequences include hypertriglyceridaemia, which can be severe, and hepatic steatosis, which can progress to cirrhosis and death from hepatic failure. For reasons as yet unexplained, affected subjects also frequently develop a hypertrophic cardiomyopathy that can lead to death from cardiac failure. Acanthosis nigricans, acromegaloid features, and muscle hypertrophy are also found.4–7
The high incidence of parental consanguinity and high recurrence rate among sibs has long suggested that BSCL is usually an autosomal recessive condition. Studies in Norwegian patients suggested a founder effect traced back to the fourteenth century.8 An initial genome wide scan performed in a multi-ethnic set of consanguineous families failed to identify a locus displaying significant homozygosity by descent.9 Subsequently, Garg et al10 identified a gene on chromosome 9q34 (BSCL1) that was linked to the disease in 17 of 19 families, mostly of African-American origin. Magré et al11 reported that this locus was unlikely to be responsible for the majority of affected families in other ethnic groups and, using extensive family material from Norway and the Lebanon, identified a second locus on 11q13 (BSCL2).11 In the 11q13 linked families, multiple different homozygous and compound heterozygous mutations were found in a previously undescribed gene, termed seipin.
The seipin gene encodes a protein of unknown function with little homology to known proteins. Surprisingly, this gene is widely expressed, with particularly high expression levels in the brain. Intellectual impairment has frequently been mentioned in more than 100 published clinical (case) reports of BSCL, but has often been attributed to factors such as the non-specific effects of chronic illness in childhood and/or the adverse social consequences of the very abnormal appearance of these children. The large number of affected subjects in the international consortium, which collaborated to identify the BSCL2 gene, provided us with an opportunity to examine the genotype/phenotype relationship in this disease.
MATERIALS AND METHODS
Description of cohort
All studies were performed with the informed consent of the subjects and/or their recognised guardians and with the formal approval of local institutional ethics committees. Patients included in the cohort presented with lipoatrophy, hyperlipidaemia (in the vast majority of cases, hypertriglyceridaemia), hepatomegaly, acromegaloid features, and insulin resistance. Seventy affected subjects from 44 independent pedigrees were studied. The families originated from a wide range of ethnicities and geographical locations (Appendix 2).
Genotyping
BSCL families were classified as BSCL1, BSCL2, or BSCLX on the basis of mutational analysis and haplotype analysis. All subjects classified as BSCL2 had homozygous or compound heterozygous mutations in the seipin gene. Subjects were classified as BSCL1 if there was evidence for cosegregation with chromosome 9q, but not with 11q. Two families showed evidence against cosegregation with either 9q or 11q and were termed BSCLX. One family was unclassifiable as no seipin mutation was found and haplotype analysis at chromosome 9 was uninformative.
Phenotypic evaluation
Each participant was evaluated by history, physical examination, and review of medical records. Participating investigators completed a standardised 98 item questionnaire. A formal assessment of intelligence quotient (IQ) was available only for a minority. Therefore, a simplified scale, based largely on educational performance, was used to assess whether there was mild, moderate, or severe intellectual impairment (Appendix 1).
Statistical analysis
From all 70 affected subjects, we considered only index cases (n=44) for statistical analysis while the remaining patients (n=26) were included in a group of relatives. Patients genotyped as BSCL2 (n=24) displayed evidence for mutations in the seipin gene on chromosome 11q. All other index cases with lipodystrophy (n=20) were considered as a unique group (BSCLs). Evidence provided for cosegregation with chromosome 9q allowed further classification as BSCL1 (n=17). Index patients lacking evidence for cosegregation with chromosomes 11q or 9q were classified as BSCLX (n=3). One patient was unclassifiable and, as such, uninformative. Congenital lipoatrophy (presence of lipoatrophy at birth) was introduced as a nominal (bimodal) variable whereas delayed lipoatrophy was introduced as a numerical variable as a function of age (month). Similarly, diabetes mellitus was introduced as a nominal variable and age of onset, when specified, was considered as a numerical variable (years). Birth weight was considered as a numerical variable (g). However, hypotheses were tested for the presence of defect in birth weight and, in this case, nominal variables were considered as defects defined under the 25th or 10th centile. Mental impairment was introduced either as a bimodal nominal variable or as a scoring index with four levels (no defect, mild, moderate, and severe). Ethnic origin (African/white) and consanguinity were introduced as bimodal nominal variables. Nominal and numerical variables were analysed with chi-square and non-parametric Mann-Whitney tests, respectively. Statistically significant values were considered at p<0.05. For the multivariate analysis, logistic regression was performed with a plurimodal equation as provided by StatView 5.0 software (Abacus Concepts, Berkeley, CA). Significant values (p<0.1) were introduced into the model equation, except data for genotyping (used for classification of groups). In the first step we compared the BSCL2 group against all other lipodystrophies but distinct groups of BSCL1 and BSCLX were also tested. Data in tables are expressed as prevalence rates (%) or as mean (SEM) in different groups of index cases and relatives.
RESULTS
Genotype
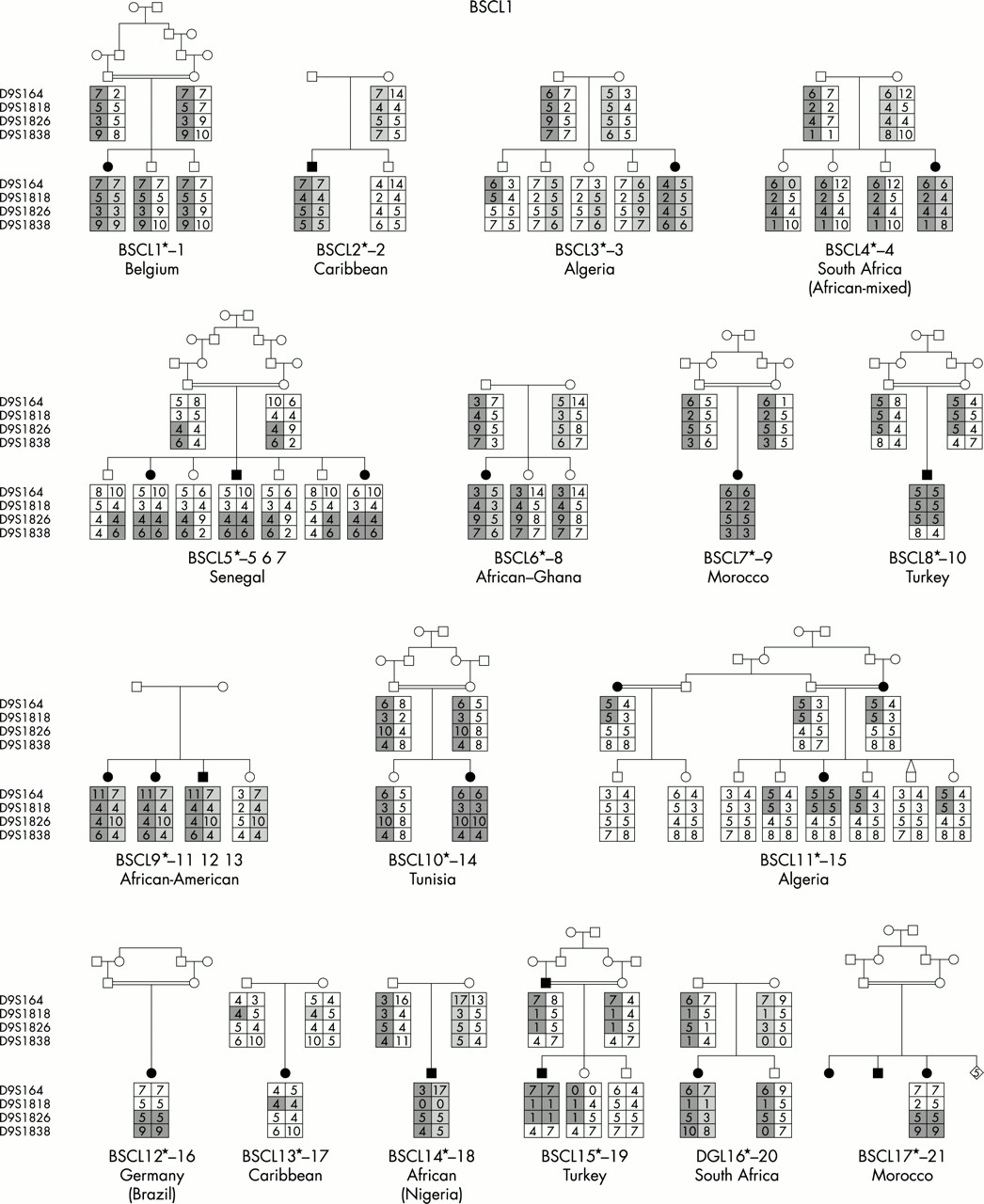
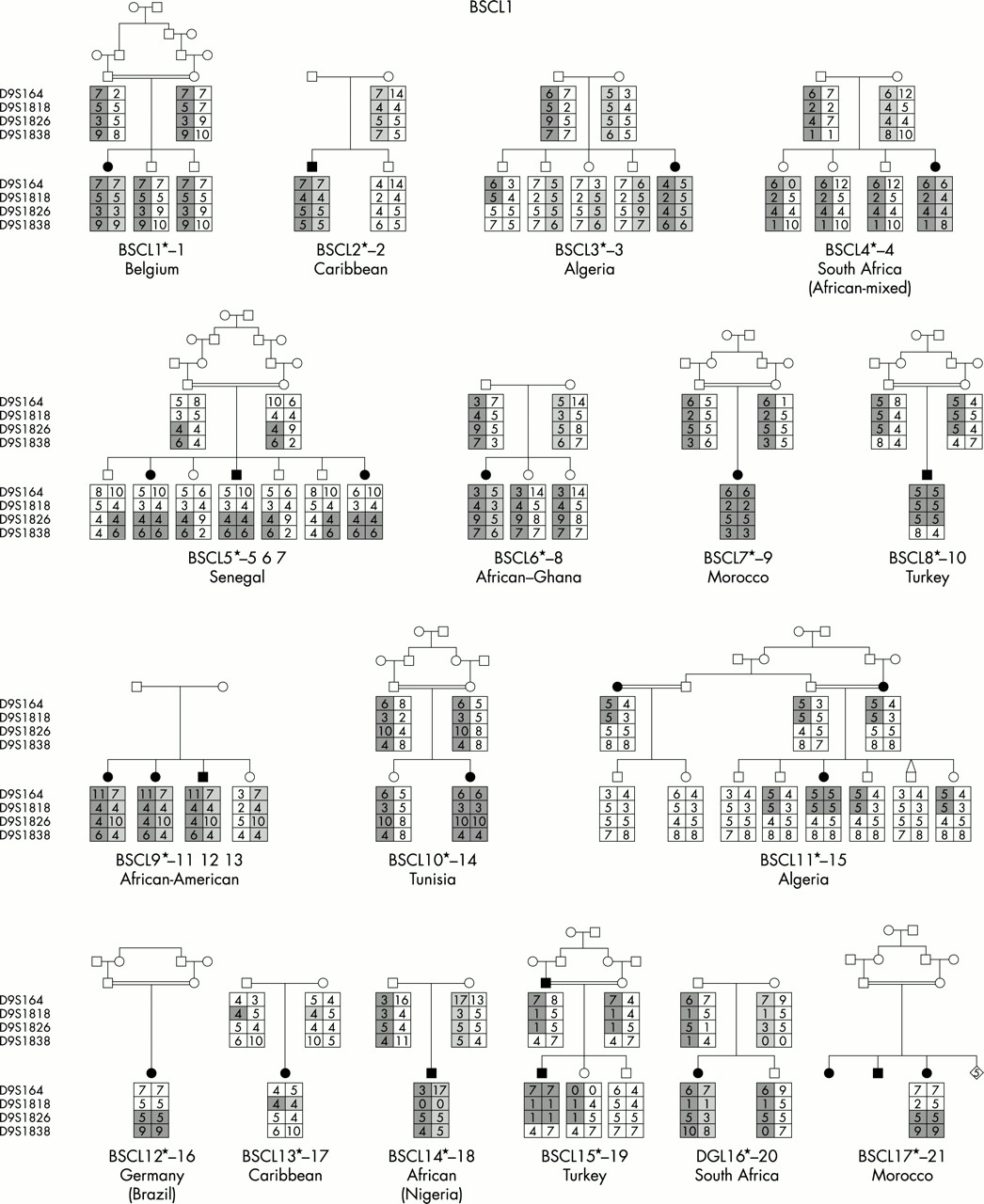
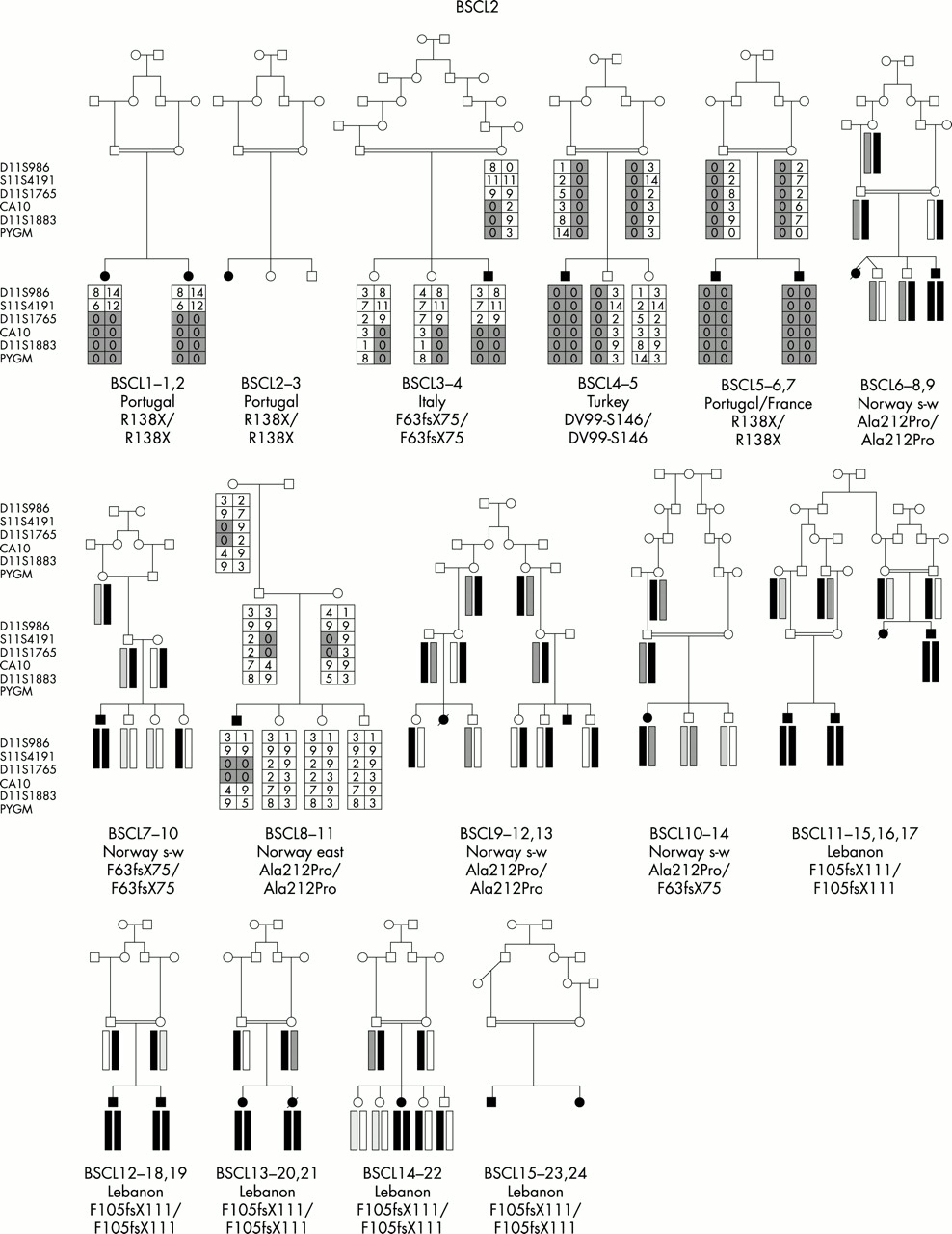
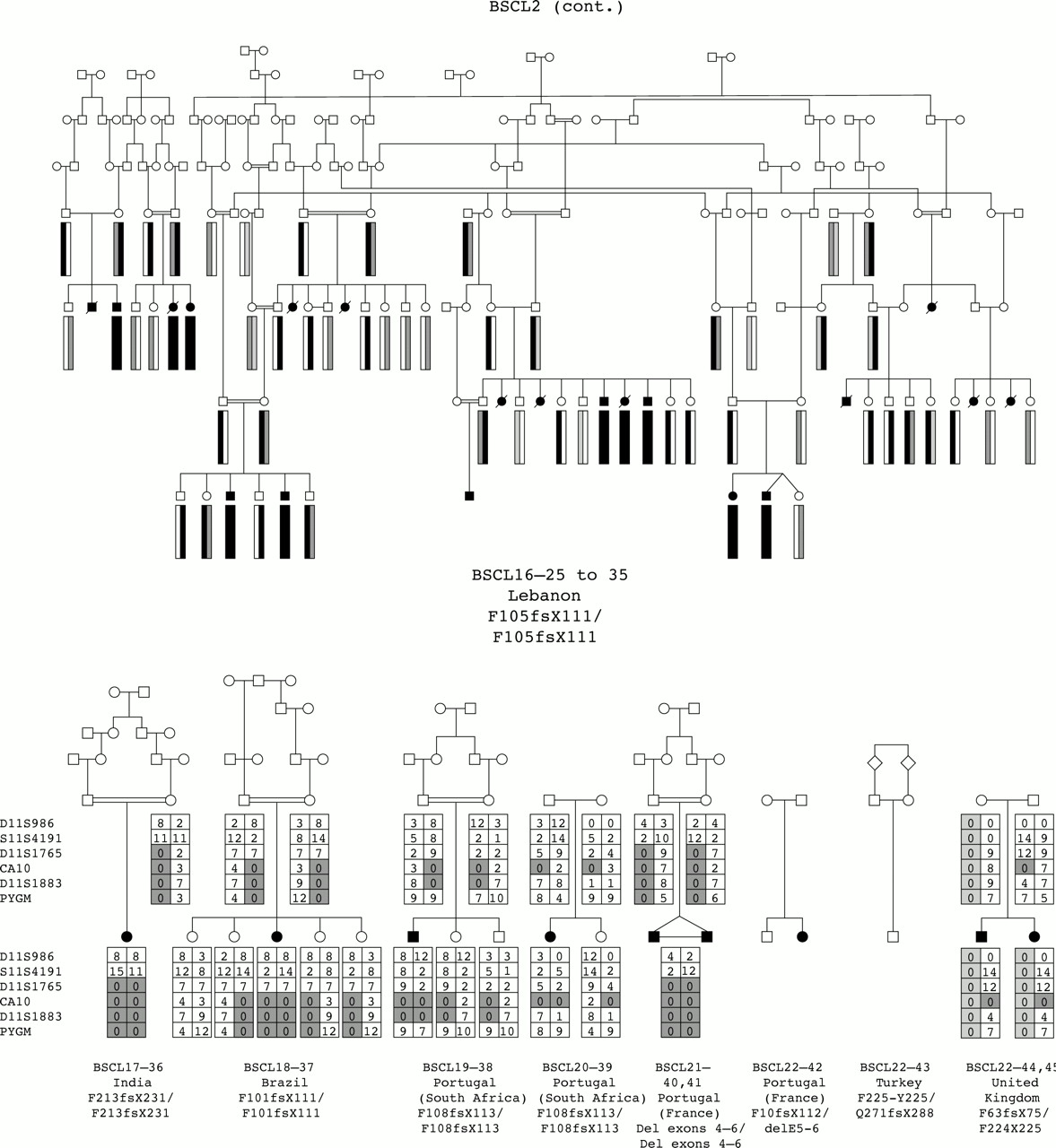
The molecular genetic analyses established that, of the total 44 families, the majority (24 families) represented BSCL2 (45 affected subjects). BSCL1 was the next largest subgroup with 17 families (21 affected subjects). One family was classified as BSCLX (one affected subject) and two families (three subjects) were unclassifiable. Appendix 2 shows the ethnic origin (country) of families within the three subtypes. Consistent with the previous findings of Garg et al,10 all families of African ancestry were in the BSCL1 group. Seipin mutations (BSCL2) were found in predominantly white subjects from Lebanon, Portugal, and Norway. The two BSCLX families as well as the non-informative family were of white European extraction. The prevalence of parental consanguinity was greater, but not significantly so, in BSCL2 than in BSCL1 or other types of lipodystrophy (table 1).
Clinical features of patients with lipodystrophy. Group 1 (BSCL2) was defined by the presence of mutations in the seipin gene, while group 2 contains all other types of BSCL. Putative linkage on chromosome 9 defined group 3 (BSCL1). Nominal variables are expressed as prevalence rate (%) and were analysed by chi-square test. Numerical variables are expressed as mean (SEM) and significance was calculated by Mann-Whitney test. Statistical significance was considered at p<0.05 comparing group 1 with groups 2 or 3.
Statistically significant differences were found when the BSCL2 group was compared to the putative BSCL1 genotype. The clinical profile of relatives of the two major BSCL types was very similar to the index cases. Ethnic differences were evident since 100% of BSCL2 cases were white, compared to only 65% in other lipodystrophies (p<0.0016). Interestingly, all (35%) subjects of African ancestry were genotyped as putative BSCL1. Further detailed study of ethnic origin indicated that, among BSCL2 subjects, 24% were from Lebanon and 56% were of Portuguese origin (p<0.005).
Phenotype
Major phenotypic features of all three subgroups taking into account index cases (used for statistical analysis) and relatives are summarised in table 1. Several major features appeared to characterise the BSCL2 group either compared to all cases of lipodystrophy or particularly to BSCL1 index cases. Thus, BSCL2 was characterised by a greater number of affected males than females, white ancestry, and highly significantly more frequent intellectual impairment. These features were also found when one considered all studied cases (index and relatives) of BSCL2 (n=45) compared to other lipodystrophies (n=25). Thus, 60% of BSCL2 patients were males compared to only 28% of other lipodystrophy cases.
Lipodystrophy and metabolic dysregulation
The majority of subjects were noted to have complete lipodystrophy either at or close to the time of birth. The prevalence of congenital onset of lipoatrophy was 79.5% in BSCL2 compared to 61% in other cases. Four subjects had a reliable history of a delayed onset of lipodystrophy and all of these were in the BSCL1 group. Although the congenital lipoatrophy was not significantly different among groups, it should be noted that the occurrence of lipoatrophy within the first year of life was 91% in BSCL2 patients compared to only 83% in other types of BSCL. There was a history of relative sparing of the face from the lipodystrophy in BSCL1 subjects of African origin, at least during the first decade of life. Hypertriglyceridaemia was reported in all subjects with marked variation in severity, even within individual families (for example, between 3 and 90 g/dl). The prevalence of diabetes mellitus was similar between BSCL2 (35.5%), BSCL1 (28.6%), and BSCLX (25%). The mean age at onset of clinical diabetes was also similar in BSCL2 and BSCL1 (17.9 (SD 1.9) and 16.3 (SD 3.4) years, respectively), whereas the age of onset for the single BSCLX patient with diabetes was 2 years. Hepatomegaly was noted in all subjects in this study.
Premature mortality
Seven of the 45 (15.5%) subjects in the BSCL2 group died prematurely between 14 and 35 years. Three of these deaths were the result of cardiac failure, two of renal failure, one of hepatic failure, and one of unknown cause. The non-informative subject died at the age of 19 months of cardiac failure. In contrast, all affected subjects in the BSCL1 pedigrees are still alive. Although this provides some evidence for a possible locus specific effect on premature mortality, the difference between BSCL2 and BSCL1/X/non-informative subgroups was not statistically significant.
Other clinical features
Skeletal muscular hypertrophy was reported in all affected subjects. The reported prevalence of hypertrophic cardiomyopathy was similar in BSCL1 (19%), BSCL2 (22%), and BSCLX (25%, including the non-informative patient), although the ability to make this diagnosis is highly dependent on access to echocardiography. While the documentation of bone cysts is dependent on patients having undergone appropriate radiological examination, it is worth noting that the frequency of bone cysts was increased in BSCL1 index patients (23.5%) compared to BSCL2 (8.3%).
Intellectual impairment
Questionnaire based information regarding intellectual performance was available for BSCL1, BSCL2, and BSCLX subjects. The reported prevalence of mild or moderate intellectual impairment was remarkably high in the BSCL2 group with 78% of subjects being affected (table 2). In contrast, only 10% of the BSCL1 subgroup and the non-informative subject were reported to show evidence of intellectual impairment. The intellectual impairment found in BSCL2 was mild in 23 subjects, moderate in 11, and severe in one subject. Of the two affected BSCL1 subjects, one was mildly and the other moderately intellectually impaired. Table 2 indicates the spectrum of mutations in BSCL2 patients grouped according to the presence or absence of intellectual impairment. There was no obvious genotype-phenotype relationship. Missense, nonsense, and frameshift mutations were equally distributed between the intellectually impaired and the normal intelligence groups. Moreover, the same nonsense mutation (F108fsX113) was observed in two unrelated subjects, of whom one was mentally retarded and the other of normal intelligence.
Seipin mutations in mentally retarded subjects and non-mentally retarded subjects
Criteria for the classification of intellectual impairment
Clinical features of all subjects listed individually
When multivariate analysis was performed by logistic regression none of the aforementioned clinical features appeared significant, except intellectual impairment. Thus, intellectual impairment as a bimodal variable was associated with BSCL2 with p<0.0001, OR 17.0, CI 3.6 to 79.0 with respect to all other types of lipodystrophies. If one compares the BSCL2 group with the BSCL1 group, again intellectual impairment appeared strongly associated with the BSCL2 genotype (p<0.0001, OR 23.5, CI 3.9 to 128.3).
DISCUSSION
We have undertaken the most comprehensive study reported to date of genotype/phenotype relationships in inherited human syndromes of congenital generalised lipodystrophy (CGL) involving 70 affected subjects from 44 independent pedigrees (fig 1). Clinically relevant findings resulting from these studies include (1) the confirmation of the existence of at least one additional CGL locus other than those on chromosomes 9 and 11, (2) the confirmation of ethnic differences in the relative frequency of BSCL1 and BSCL2 gene defects, and (3) the establishment of certain key phenotypic differences, in particular concerning intellectual impairment, between the three subtypes of CGL.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Family pedigrees of all participating families grouped by aetiology. For BSCL1 families, evidence for cosegregation with 9q34 is provided. For BSCL2, the nature of the mutation identified is indicated under the pedigree. For BSCLX, evidence against cosegregation with the 9q34 locus is also provided.
Two Europid families in our cohort had no detectable seipin mutations and also showed evidence against the involvement of a locus at 9q34. Taken together with the data of Heathcote et al12 in Omani pedigrees, this strongly suggests that there must be at least one additional locus that causes BSCL. For the moment, we suggest that this be referred to as BSCLX.
Among the 44 families studied, BSCL1 patients originated primarily from sub-Saharan Africa and Maghreb countries (Morocco, Algeria, Tunisia) with only a minority coming from Middle Eastern countries (for example, Turkey) and northern Europe. This is consistent with the results of Garg et al,10 who found cosegregation of the disease with chromosome 9q34 markers in the great majority of their families of African-American origin. In contrast, a large number of different seipin (BSCL2) mutations were found in subjects of varying white ethnicities including Norway, United Kingdom, and Mediterranean countries, as well as Middle Eastern Arabs. Interestingly, patients from Portugal and its ancient colonies accounted for a significant proportion of subjects, suggesting a spread from the 15th century onwards.13 Notably, no subjects of African ancestry had seipin mutations. The two BSCLX families as well as the non-informative patient were of Europid origin.
The higher prevalence of recorded inbreeding in BSCL2 families than in BSCL1 families cannot easily be evaluated as long as the various local inbreeding patterns are not known. For example, chance inbreeding has been verified in one Norwegian BSCL2 genetic compound family. Taking into account also the different ethnic origins, the simplest explanation is that the high African origin of BSCL1 is the result of a single, very ancient mutation having reached an appreciable spread and allele frequency.
Phenotypic evaluation was undertaken by multiple independent physicians in several countries and patients were not examined by a single observer. Since a standardised formal assessment of intellectual capabilities was difficult to establish because of highly variable attendance at school and the wide range of ethnic and racial background, it was decided to evaluate the degree of mental impairment using a grow scale measuring social adaptation and school performance. This classification allows some overlap between severe and moderate mental retardation.
Notwithstanding these caveats, the application of standardised questionnaires across these multiple centres has allowed us to attempt to establish, with reasonable confidence, whether there are any major and consistent phenotypic differences between the genetic subtypes of this disease. Lipoatrophy was present at birth and complete in all BSCL2 and BSCLX subjects. In contrast, a subset of BSCL1 subjects had evidence for lipoatrophy that was either delayed, incomplete, or both. For those subjects in whom the lipoatrophy was incomplete, the face tended to be spared. Thus, BSCL1 may show phenotypic overlap with at least two other forms of lipodystrophy that have previously been considered distinct. Acquired total lipodystrophy (Lawrence syndrome) is generally considered to be a sporadic, non-inherited condition in which total lipodystrophy, of presumed autoimmune origin, occurs often after an infectious illness.14 Our findings suggest that some cases of BSCL1 in which the onset of the lipodystrophy is delayed and are of sporadic occurrence may be labelled incorrectly as having Lawrence syndrome. The distinction is made more complex by the fact that there are no diagnostic tests for Lawrence syndrome, although the lipoatrophy of this syndrome does tend to appear more suddenly than in the BSCL1 cases. Familial partial lipodystrophy of the Dunnigan-Köberling type (FPLD, OMIM 151660) is an autosomal dominant, face sparing lipodystrophy caused by a particular cluster of mutations in the nuclear envelope proteins lamins A/C.15–19 However, these patients have a distinctive cushingoid appearance of the face that makes differential diagnosis easy.
The relative effects of the BSCL subtypes on premature mortality are difficult to quantify given factors such as incomplete ascertainment in families and possible referral bias. However, it is notable that while none of the BSCL1 patients included in our study died before the age of 40 (but family history death of affected subjects in early adulthood was positive in some cases), premature death was observed in seven patients from the BSCL2 subgroup. The reasons for this difference are not obvious as the prevalence of major metabolic derangements (for example, diabetes, hyperlipidaemia, and hepatic dysfunction) were similar between the groups. However, it is notable that three of the seven premature deaths in the BSCL2 group were attributed to cardiac failure, including one during pregnancy.
As these diseases are clearly autosomal recessive it was surprising that we found an excess of females in the BSCL1 group and an excess of males in the BSCL2 group. In the case of BSCL1, this is most likely to represent ascertainment bias, as the appearance of lipodystrophy is often more striking in females leading to earlier referral and diagnosis. However, this would not explain the opposite sex ratio found in the BSCL2 group. Within that group, affected female subjects tended to have a more severe phenotype than males (personal observations) and this subtype is generally more lethal than BSCL1. It is possible, therefore, that earlier mortality in affected females has resulted in a systematic bias towards the referral of surviving males to the consortium.
Interestingly, radiological evidence of bone cysts was observed in 10 patients (five with BSCL1, three with BSCL2, and two with BSCLX). A syndrome of cystic angiomatosis of bones and congenital lipodystrophy (OMIM 272500) has been reported by Brunzell et al20 in 1968 and considered to be an entity separate from BSCL. Our findings suggest that this may not be a discrete entity.
The most remarkable difference between the groups was the prevalence of intellectual impairment. While it was not possible to undertake thorough and systematic quantitative assessment of IQ, it is notable that while 78% of BSCL2 subjects were reported as having some degree of intellectual impairment, this was true of only 10% of the BSCL1 subjects. Although this difference might be explained by the expression of seipin in the brain, such a tissue specific expression may also be independent from any type of mental retardation (Wolfram syndrome, Duchenne muscular dystrophy, merosin positive congenital muscular dystrophy) and does not therefore provide a definitive clue to its pathophysiology. The normal brain MRI reported in mentally retarded BSCL2 patients is also remarkable.
No clear correlation was apparent between the site and type of seipin mutation and intellectual impairment. Furthermore, affected subjects from different families, but harbouring the same mutation, were found to be discordant in terms of mental impairment in some instances. Clearly, given the complex and qualitative nature of cognitive functions, it is not surprising that its expression may be subject to multiple genetic and environmental modifiers.
In summary, BSCL1 is a form of congenital lipodystrophy, most prevalent among subjects of African origin, which appears to be a milder disease than BSCL2. This is manifested by its more frequent presentation as a delayed or partial lipodystrophy, a lower occurrence of premature death, and a markedly lower incidence of intellectual impairment. These observations suggest that the causative gene in BSCL1, yet to be described, may turn out to have more restricted functions and/or a more limited expression pattern than seipin. Our findings emphasis the importance of attempting to make a molecular diagnosis of the syndromes of congenital lipodystrophy as this may aid both prognostication and genetic counselling.
Acknowledgments
We thank the patients, their families, and the referring physicians for their participation; A Lemainque, P Hilbert, L Bodson, F Cavallin, A Fauré, V La Villa, and S Pavek for genotyping; and D Recan and her colleagues for immortalising the lymphoblasts. We thank the rare diseases programme of the European Commission (Directorate-General XII) for its support and L’Aide aux Jeunes diabétiques, l’Association Française contre les Myopathies (AFM), the Anders Jahres Fund, Assistance Publique-Hôpitaux de Paris, INSERM, Medinnova, and Ministére de la Recherche for their constant encouragement and financial support. We also want to thank Christian Melot and Maurice Jottrand for their help in statistical analysis of the data.
NOTE ADDED IN PROOF After submission of this paper, identification of the BSCLI gene was published by Agarwal A, et al (Nat Genet 2002;31:21–3). The AGPAT2 gene encodes a key enzyme in the biosynthesis of triacylglycerol. This is consistent wth the limited expression pattern hypothesised in our conclusion.
REFERENCES
Linked Articles
- Correction