Article Text

Abstract
Beckwith-Wiedemann syndrome (BWS) is a model imprinting disorder resulting from mutations or epigenetic events involving imprinted genes at chromosome 11p15.5. Thus, germline mutations inCDKN1C, uniparental disomy (UPD), and loss of imprinting of IGF2 and other imprinted genes have been implicated. Many familial BWS cases have germlineCDKN1C mutations. However, most BWS cases are sporadic and UPD or putative imprinting errors predominate in this group. We have identified previously a subgroup of sporadic cases with loss of imprinting (LOI) of IGF2 and epigenetic silencing of H19 proposed to be caused by a defect in a distal 11p15.5 imprinting control element (designated BWSIC1). However, many sporadic BWS patients show biallelicIGF2 expression in the presence of normalH19 methylation and expression patterns. This and other evidence suggested the existence of a further imprinting control element (BWSIC2) at 11p15.5. Recently, we showed that a subgroup of BWS patients have loss of methylation (LOM) at a differentially methylated region (KvDMR1) within theKCNQ1 gene centromeric to theIGF2 and H19genes. We have now analysed a large series of sporadic cases to define the frequency and phenotypic correlates of epigenetic abnormalities in BWS. LOM at KvDMR1 was detected by Southern analysis or a novel PCR based method in 35 of 69 (51%) sporadic BWS without UPD. LOM at KvDMR1 was often, but not invariably associated with LOI ofIGF2. KvDMR1 LOM was not detected in BWS patients with putative BWSIC1 defects and cases with KvDMR1 LOM (that is, putative BWSIC2 defects) invariably had a normalH19 methylation pattern. The incidence of exomphalos in putative BWSIC2 defect patients was not significantly different from that in patients with germlineCDKN1C mutations (20/29 and 13/15 respectively), but was significantly greater than that in patients with putative BWSIC1 defects (0/5, p=0.007) and UPD (0/22, p<0.0001). These findings are consistent with the hypothesis that LOM of KvDMR1 (BWSIC2 defect) results in epigenetic silencing ofCDKN1C and variable LOI ofIGF2. BWS patients with embryonal tumours have UPD or a BWSIC1 defect but not LOM of KvDMR1. This study has further shown how (1) variations in phenotypic expression of BWS may be linked to specific molecular subgroups and (2) molecular analysis of BWS can provide insights into mechanisms of imprinting regulation.
- Beckwith-Wiedemann syndrome
- epigenotype-phenotype correlations
- imprinting
Statistics from Altmetric.com
Beckwith-Wiedemann syndrome is a prototypic imprinting disorder resulting from mutations or epigenetic alterations (epimutations) in genes from an imprinted cluster at chromosome 11p15.5.1 2Phenotypic expression of BWS is variable, but the major features are pre-/postnatal overgrowth, macroglossia, and abdominal wall defects.3 Additional features include organomegaly, hypoglycaemia, hemihypertrophy, genitourinary abnormalities, and a susceptibility to embryonic tumours which occur in 5% of patients. The genetics of BWS are complex,1 2 but three main groups are distinguished: ∼15% have familial BWS, ∼2% have cytogenetic abnormalities (duplications or balanced rearrangements) involving chromosome 11p15.5, and the rest are sporadic. Imprinted genes have been implicated in the pathogenesis of both familial and sporadic BWS.
Genomic imprinting is an epigenetic mechanism in which gene expression is altered according to the parental origin of the allele. It is estimated that the human genome may contain between 100 and 1000 imprinted genes and most of those identified to date are arranged in clusters. A number of imprinted genes from chromosome 11p15.5 have been implicated in the pathogenesis of BWS including the maternally expressed CDKN1C,KCNQ1, and H19genes and the paternally expressed IGF2 andKCNQ1OT genes. The molecular pathology of familial and sporadic BWS differs. Thus, although germline mutations in the cyclin dependent kinase inhibitor geneCDKN1C may be detected in >40% of familial cases, CDKN1C mutations are infrequent (<5%) in sporadic cases.4-9 Among sporadic cases, ∼20% have uniparental disomy (mosaic paternal isodisomy) for the 11p15.5 imprinted gene cluster.10-12 In addition, many sporadic cases show loss of imprinting (LOI) ofIGF2 resulting in biallelic expression.13-15
The epigenetic alterations associated withIGF2 LOI are heterogeneous. Thus, we described initially a subgroup of sporadic patients with biallelicIGF2 expression and silencing ofH19 expression in which there wasH19 hypermethylation such that the maternalH19 allele showed a paternal epigenotype with H19 promoter methylation.12 14 Subsequently, we showed that in many sporadic cases IGF2 LOI occurs via anH19 independent mechanism.15These findings suggested that chromosome 11p15.5 contained two imprinting control centres/elements termed BWSIC1 and BWSIC2. Mutations or epimutations at BWSIC1 would result inIGF2 LOI and H19silencing, while BWSIC2 defects would be associated withIGF2 LOI through anH19 independent mechanism.
Investigations of the recipricosity of IGF2and H19 imprinting in mice suggested that BWSIC1 was likely to map to the H19 upstream region. The finding of H19 independent LOI of IGF2 in a BWS patient with a chromosomal rearrangement disruptingKCNQ1 16 suggested that BWSIC2 mapped centromeric to IGF2 close to (or within) KCNQ1. Recently, a differentially methylated region (KvDMR1) within KCNQ1 was identified.17-19 KvDMR1 shows maternal allelic methylation and the unmethylated paternal allele expresses an antisense RNA (KCNQ1OT (also known asLIT1)). We observed that some BWS patients showed loss of methylation at KvDMR1 and (for those that were informative) this was associated with H19independent IGF2 LOI (consistent with a BWSIC2 defect).17
Previously, we reported significant correlations between the phenotypic variability and molecular pathology in BWS. Thus, exomphalos was not found in BWS patients with UPD or BWSIC1 defects, but was present in the majority of patients with germlineCDKN1C mutations.9 12 We now report further investigations to define the role of LOM of KvDMR1 (BWSIC2 defects) in sporadic BWS and to elucidate epigenotype-phenotype correlations.
Patients and methods
BWS PATIENTS
A diagnosis of BWS was made according to our standard diagnostic criteria; either (1) three major features (anterior abdominal wall defects, macroglossia, and pre-/postnatal growth >90th centile) or (2) two major plus three or more minor features (characteristic ear signs (ear lobe creases or posterior helical ear pits), facial naevus flammeus, hypoglycaemia, nephromegaly, and hemihypertrophy).3 Patients with normal chromosomes and no evidence of UPD were analysed for KvDMR1 methylation status and germline CDKN1C mutations as described below. Uniparental disomy analysis and H19methylation analysis was performed as reported previously.9 13 14
MOLECULAR GENETIC ANALYSIS
KvDMR1 methylation analysis
Two approaches were used to determine methylation status at KvDMR1. Firstly, Southern analysis was performed on genomic DNA isolated from whole blood as described previously.17Southern analysis was performed in nine new cases (eight further cases have been reported previously.17 However, for many BWS patients insufficient DNA was available for Southern analysis and a PCR based approach was developed to identify patients with loss of KvDMR1 methylation. DNA (500 ng) (isolated from whole blood) was digested overnight with the methylation sensitive enzymeNotI (New England Biolabs) according to the manufacturer's instructions. It was then ethanol precipitated, resuspended in 5 μl of sterile distilled water, and the whole sample subjected to multiplex PCR as set out below. A total of 100 ng of undigested DNA for each sample was PCR amplified in exactly the same way at the same time to act as a normal control for comparison with the digested sample.
Multiplex PCR was carried out using two sets of primers, which were designed using sequence information from PAC DJ74K15. Set 1 crosses the methylation sensitive NotI site, KvLF1-FAM-AGACCTTGCCCGG GTT, KvLR1–AAAGTGATGTGCCGTGT CCT and set 2 acted as a control reaction amplifying an upstream region, KvLF2-HEX-CCCATCTGCACCTTATGGAC, KvLR2-CCAGGATGTGGACCCTAGAC. The forward primer for each set was tagged with a different fluorescent dye.
Thirty μl PCR reactions were set up containing 6.6 pmol of each primer, 2 μl dNTPs (2.5 mmol/l), 1.5 mmol/l Mg, 10% DMSO, buffer, and 0.2 μl Taq DNA polymerase. Reactions were overlaid with mineral oil and cycled at the following conditions with the hotlid on: 95°C for 10 minutes once (this step allows for an eight minute hotstart), 94°C, 66°C, 72°C for 30 seconds each ×3, 94°C, 64°C, 72°C for 30 seconds each ×3, 94°C, 62°C, 72°C for 30 seconds each ×3, 94°C, 56°C, 72°C for 30 seconds each ×20.
All samples were separated and analysed using the Genescan software (ABI). Values for the peak heights for each PCR product were taken and used to calculate a single ratio. This ratio measures the reduction in methylation sensitive PCR product after digestion, and is therefore an indication of the levels of methylation in the sample. A methylation ratio of 0.1 or less was chosen as the value indicative of loss of methylation at this site. Patient samples that fell into this category were verified by repetition; if the low reading could not be repeated twice in three tests it was disregarded.
CDKN1C mutation analysis
We have previously reported CDKN1Cmutation analysis in 54 sporadic BWS patients.9 To define the molecular pathology of BWS further, we extended this analysis by analysing a further 30 sporadic cases using the methods described previously.9
GENOTYPE-PHENOTYPE ANALYSIS
To compare the phenotype of different subgroups of BWS defined by molecular genetic analysis, we compared those patients with loss of methylation (LOM) at KvDMR1 identified in this study with (1) 22 patients with UPD, nine of whom were reported previously,9 12 (2) five patients withH19 hypermethylation reported previously,12 14 and (3) 15 patients with germlineCDKN1C mutation (13 of whom were reported previously9 and two identified in this study). Statistical comparisons were made using Fisher's exact test and statistical significance was set at 0.05.
Results
KvDMR1 METHYLATION ANALYSIS
KvDMR methylation status was examined by Southern analysis or a novel PCR based assay or both in 69 patients referred with BWS in whom a cytogenetic rearrangement and uniparental disomy (UPD) had been excluded.
Southern analysis was performed in 17 patients (eight reported previously). Loss of KvDMR1 methylation was detected in eight cases (three reported previously17). However, in many further cases insufficient DNA was available to perform KvDMR1 methylation analysis using this approach. We therefore developed a novel PCR based technique to detect patients with LOM at KvDMR1 (see Methods).
To validate our PCR based strategy for detecting KvDMR1 LOM, we analysed DNA from 66 normal controls and confirmed that none gave a positive result. We then analysed 67 patients with confirmed or suspected BWS using the PCR based assay. A positive result (indicating loss of KvDMR1 methylation) was obtained in 35 cases (fig 1). KvDMR1 methylation status was analysed by both Southern and PCR based strategies in 15 patients and there was complete concordance between the two approaches.
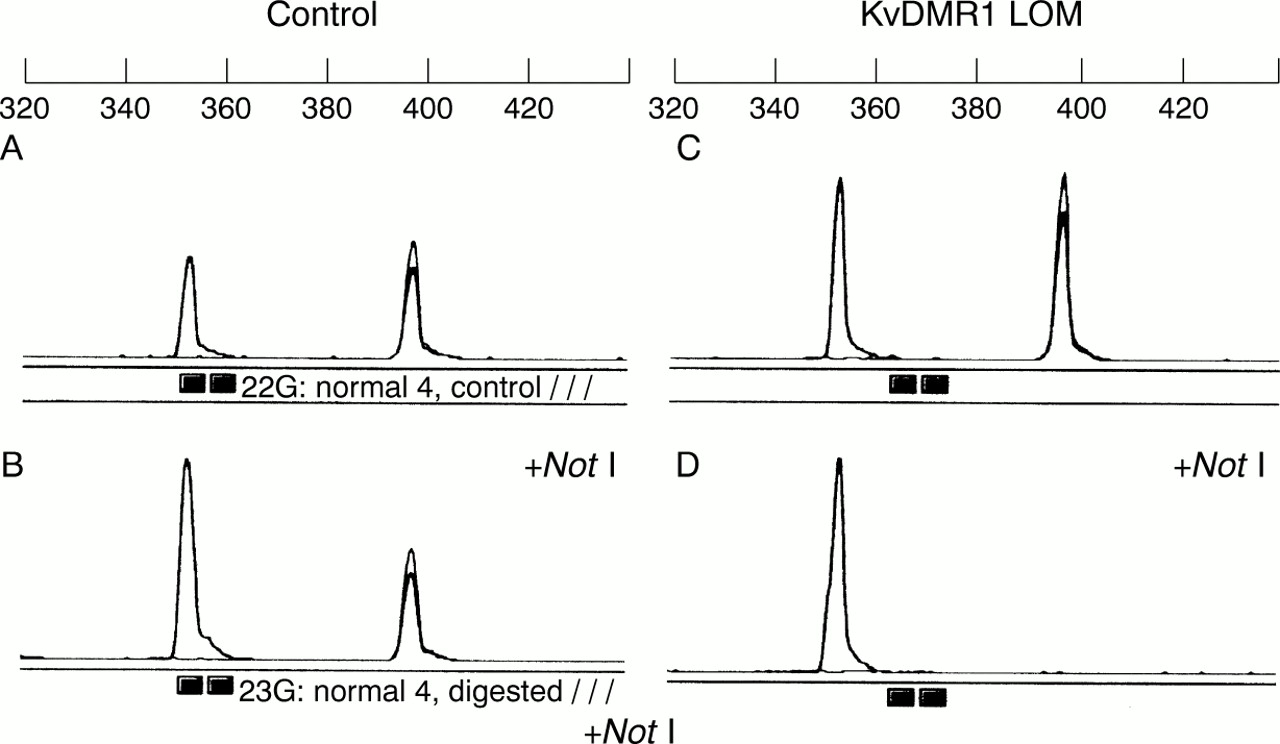
Detection of loss of methylation (LOM) at KvDMR1 by a PCR based assay. Multiplex PCR of control DNA shows two PCR products at ∼350 and ∼400 bp (see panel A). Multiplex PCR of control DNA following digestion with NotI. The ∼400 bp product is reduced to ∼50% by NotI digestion of the unmethylated paternal allele (see panel B). Panels C and D show results for a BWS patient. Panel C shows two PCR products similar to those for control DNA (panel A). However, following digestion with NotI there is no ∼400 bp PCR product as both the paternal and the unmethylated maternal KvDMR alleles are cut.
LOSS OF METHYLATION AT KvDMR1 AND IGF2/H19 EPIGENOTYPE
Thirty three of 69 patients analysed for KvDMR1 methylation by Southern or PCR based techniques had previously been analysed forH19 methylation status.12 14All of the 23 patients with KvDMR1 loss of methylation in whomH19 methylation status had been examined showed normal H19 methylation. Conversely, four patients with H19 hypermethylation (BWSIC1 defect) displayed normal KvDMR1 methylation. In a previous publication, we reported that the two patients with KvDMR1 demethylation in whom IGF2 imprinting status had been defined both showed biallelic IGF2expression.17 Using the PCR based assay, we identified two further BWS patients with LOM at KvDMR1 in whomIGF2 expression had been defined.15 However, while one patient did show loss ofIGF2 imprinting, the other retained monoallelic IGF2 expression.
GERMLINE CDKN1C MUTATION ANALYSIS
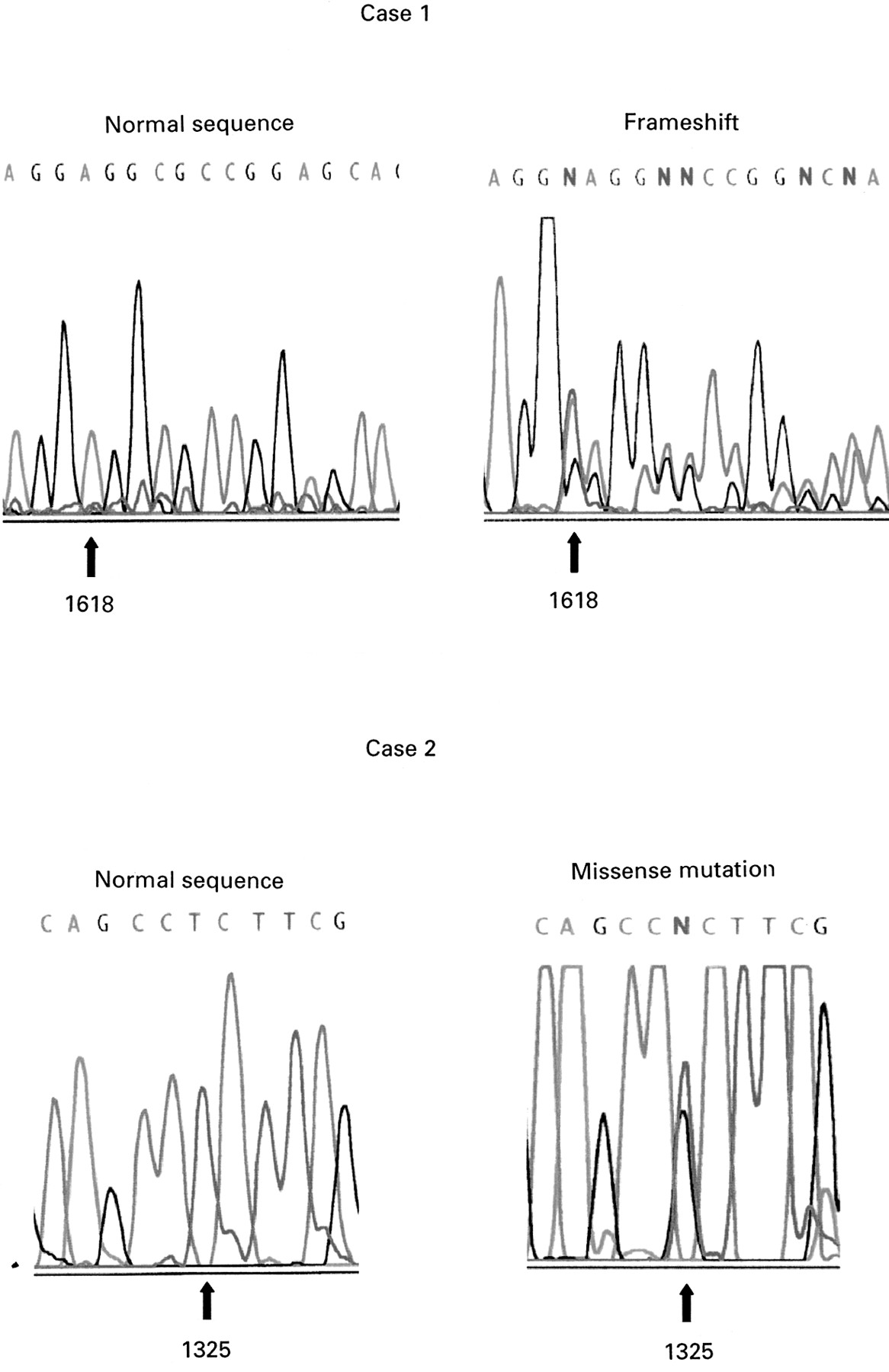
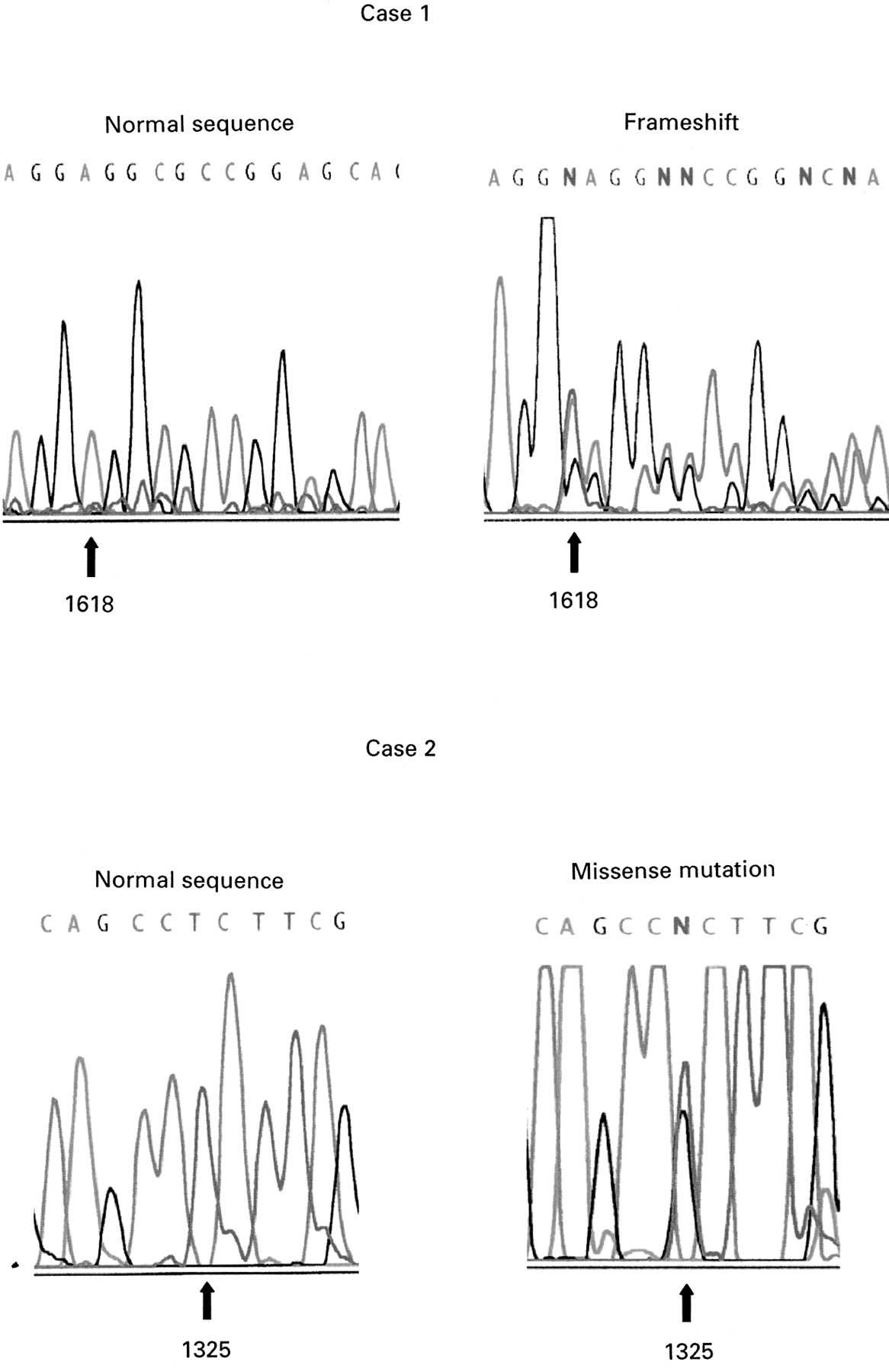
To provide further information on the frequency of germlineCDKN1C mutations in sporadic cases and to enhance genotype-phenotype comparisons (see later), we analysed a further 30 sporadic cases. Germline CDKN1Cmutations were identified in two cases (fig 2).
{kind=link}
{kind=link}
Germline CDKN1C mutations in sporadic BWS. Case 1: insertion of T at nucleotide 1618 causes a sequence frameshift. Case 2: mutation 2. T to G transversion at nucleotide 1325 produces a non-conservative leucine to arginine substitution at codon 33.
In case 1, a frameshift insertion of a T at nucleotide 1618 was predicted to produce a CDKNIC protein truncated proximal to the PAPA repeat and PCNA binding domains. The mutation was not detected in parental DNA and appeared to have arisen de novo. In case 2, a T to G transversion at nucleotide 1325 caused a non-conservative substitution of leucine to arginine at codon 33 within the cyclin binding domain. This residue is conserved in the mousecdkn1c gene. The mutation was also present in the mother but not present in paternal DNA.
GENOTYPE-PHENOTYPE CORRELATIONS
To determine possible genotype-phenotype correlations, we compared the frequency of exomphalos and embryonal tumours in patients with KvDMR1 methylation to that in other well defined molecular subgroups of BWS (see Methods). Of 29 BWS patients with LOM of KvDMR1 for which sufficient clinical information was available, 20 of 29 (69%) cases had exomphalos. Compared to other molecular subgroups, the frequency of exomphalos in patients with LOM of KvDMR1 (BWSIC2 defect) was significantly greater than for patients with BWSIC1 defects (20/29versus 0/5, p=0.007) or UPD (20/29v 0/22, p<0.0001), but was similar to that in patients with germline CDKN1C mutations (20/29 v 13/15, p=0.28). None of the patients with a LOM of KvDMR1 had developed an embryonal tumour, and BWS patients with tumours (n=3) had UPD (2/22) or a BWSIC1 defect (1/5).
Discussion
We have further investigated the molecular pathology of sporadic BWS. Combining our current and previous results forCDKN1C mutation analysis,9 we have now identified germline mutations in almost 5% (four of 84) of sporadic cases. In these patients, theCDKN1C mutation was maternally inherited in one case, arose de novo in two cases, and was of uncertain origin in one case. The identification of a maternally inheritedCDKN1C mutation in an isolated case has important implications for future pregnancies. The putative missense mutation in case 2 caused a non-conservative substitution of an evolutionarily conserved amino acid in the CDK inhibitor domain. To the best of our knowledge, a total of 14 germlineCDKN1C mutations have now been described in BWS including three missense mutations (two of which were in the CDK binding domain).4-9
The finding of KvDMR1 demethylation in a large proportion of sporadic BWS cases suggests that studies of KvDMR1 methylation and UPD status should be the initial investigations in BWS. Our PCR based assay was designed to detect patients with complete KvDMR1 LOM and patients mosaic for demethylation would not be expected to be detected so our results may provide an underestimate. We have confirmed and extended our findings that LOM at KvDMR1 is not associated with changes inH19 methylation, and that BWS patients withH19 hypermethylation and silencing do not show alterations in KvDMR1 methylation.17 These findings are consistent with the hypothesis that there are two “imprinting control elements” (or imprinting centres) within the 11p15.5 imprinted gene cluster. Thus, in humans, BWSIC1 would regulateIGF2 and H19imprinting while BWSIC2 would regulate IGF2,CDKN1C, KCNQ1, and KCNQ1OT imprinting.2 The results of transgenic mouse experiments support this model asCdkn1c and Kvlqt1(Kcnq1) imprinting appears to be independent of H19.20 In the mouse, Cdkn1c expression requires a maternal germline imprint,21 although maternal germline methylation has not yet been found in or near the gene. Transgenic experiments have failed to identify a functional imprinting signal within 35 kb surrounding the Cdkn1c gene,22suggesting that Cdkn1c imprinting might be influenced by the BWSIC2 locus.2 Indeed, in a recent publication, Horike et al 23reported that a targeted deletion of KvDMR1 (in a modified human monochromosome hybrid system) silenced paternalKCNQ1OT (LIT1) expression and activated expression of KCNQ1and CDKN1C with no effect onH19 imprinting.
The results of the current study are compatible with our previous suggestion that most sporadic BWS patients have epigenetic alterations in imprinted genes. They also confirm that BWSIC2 defects are more frequent than those at BWSIC1.12 14 15 17 24Imprinting centre defects are well defined in Prader-Willi and Angelman syndromes (PWS/AS).25-27 In these disorders the detection of germline deletions has enabled the precise localisation of the sequences involved in imprint control. To date, no equivalent germline deletions have been identified in BWS patients. In patients with putative BWSIC1 defects, no evidence of H19gene deletions or mutations have been identified.12 14 28 However, mutations could be present in upstream regulatory regions that have not yet been analysed.29 30 We note that in PWS/AS cases, epigenetic errors (epimutations) rather than mutations predominate in sporadic cases27 and BWS patients with putative BWSIC1 and BWSIC2 defects may also result from epimutational events. Although limited Southern analysis has not yet shown evidence of germline deletions in BWS patients with LOM at KvDMR1, further investigations are required.
KvDMR1 is close to the transcription initiation site of a paternally expressed antisense transcript (KCNQ1OT orLIT1). Patients with loss of methylation at KvDMR1 show biallelic expression of KCNQ1OT. It has been proposed that KvDMR1 may be a boundary element that separates CDKN1C andKCNQ1 promoters from a downstream enhancer and that the boundary is open when methylated.2 This model would predict that loss of KvDMR1 methylation would close the boundary leading to reduced expression of KCNQ1 andCDKN1C.2 Previously we had reported that two patients with KvDMR1 LOM showed biallelicIGF2 expression. These results contrasted with those of Lee et al 18 who reported monoallelic IGF2 expression in four of five patients with LOI of KCNQ1OT. In this study we identified two further BWS patients with KvDMR1 methylation in whomIGF2 imprinting status was defined. Intriguingly, one showed biallelic IGF2expression and one monoallelic IGF2expression. These results reconcile the differences between our previous findings (in which all patients with KvDMR1 LOM had biallelicIGF2 expression) and those reported by Leeet al.18 It appears that LOM of KvDMR1 is variably associated with the loss of imprinting ofIGF2. The precise reasons for this variability is of great interest and could reflect the precise nature of the mutation or epimutation causing LOM of KvDMR1. Thus, a patient with a BWSCR1 breakpoint showed H19independent LOI of IGF2,16 so the presence of IGF2 LOI may indicate a more complete defect in BWSIC2 function. The precise mechanism by which LOM at KvDMR1 would cause LOI of IGF2 is unknown. One possibility is that KvDMR1 LOM is associated with higher order chromatin modification that would alter imprinting over a large region. However, although loss of asynchrony of replication timing was observed in a BWS patients with a chromosome 11p15.5 inversion breakpoint and H19 independent LOI of IGF2,16 Squireet al 24 reported normal asynchronous replication patterns in another BWS patient with a chromosome 11 inversion and in a patient with LOI ofIGF2 and normalH19 methylation (KvDMR1 methylation was not assessed). A further possibility is that LOM of KvDMR1 results in closure of a boundary element between CDKN1Cand a telomeric enhancer, which might then allow the enhancer to interact with the maternal IGF2 allele. We note the recent report of IGF2 control regions such as DMR1 which can influenceIGF2 imprinting via aH19 independent pathway.31
BWS shows clinical variability and molecular heterogeneity. We have identified phenotypic differences between subgroups of BWS with defined molecular pathology.9 11 12 We have now extended these correlations by showing that exomphalos is frequent in patients with germline CDKN1C mutations and in patients with putative BWSIC2 defects but not in patients with UPD or BWSIC1 defects. These findings suggest that the precise BWS phenotype is determined by the loss of function of specific genes in the imprinted cluster. The association of exomphalos with germlineCDKN1C mutations but not BWSIC1 defects (these patients in contrast are predicted to haveIGF2 overexpression but no alterations inCDKN1C) is consistent with the phenotypic differences between “BWS mice” withCDKN1C inactivation orIGF2 expression.32 33 The overlapping phenotypes of BWS patients with germlineCDKN1C mutations andIGF2 overexpression (as assumed in BWSIC1 defect cases) suggest that both gene products may act in the same biochemical pathway. Although the two imprinting domain model for 11p15.5 predicts that maternal CDKN1Cexpression will be downregulated in BWSIC2 cases, to date it has not been possible to determine CDKN1C expression patterns in patients with LOM at KvDMR1. However, the high frequency of exomphalos in patients with putative BWSIC2 defects is consistent with the hypothesis that LOM at KvDMR1 will lead to reducedCDKN1C transcription. Phenotypic variability in patients with BWSIC2 defects will also be influenced by whether this is associated with LOI of IGF2.
The ability to predict which BWS patient are at risk for embryonal tumours would enhance clinical management. Although a very high incidence of Wilms and other tumours had been suggested in BWS patients with UPD, more recent studies suggest that the risk is lower than feared previously.10 28 It is of interest that although LOI of IGF2 andH19 methylation (consistent with a BWSIC1 defect) are common in Wilms tumour,34 35 loss of imprinting of KCNQ1OT(LIT1) (consistent with a BWSIC2 defect) has not been found in Wilms tumours.19 This might suggest that the risk of embryonal tumours would be less in BWS children with BWSIC2 defects (possibly related to the lack of apparentH19 silencing in these cases36). Consistent with this hypothesis, we found that embryonal tumours were confined to BWS patients with UPD and BWSIC1 defects. However, further studies are required to delineate the exact risks of embryonal tumours in different subgroups of BWS. Although overexpression of IGF2 has long been suspected as a cause for BWS, reports of LOI ofIGF2 (if this, as expected, leads to increased IGF2 protein levels) in patients with partial or no features of BWS37 38 suggest that LOI ofIGF2 per se is not sufficient to cause BWS and alterations in expression or mutations of other genes (for example,CDKN1C) may also be required for the BWS phenotype.
Acknowledgments
We thank the many colleagues who referred patients. We are grateful to The Birth Defects Foundation (EM and WR), Wellcome Trust (EM and PS), and Cancer Research Campaign and HFSP (WR) for financial support.