Article Text

Abstract
Multiple endocrine neoplasia type 2 (MEN 2) is an inherited cancer syndrome characterised by medullary thyroid carcinoma (MTC), with or without phaeochromocytoma and hyperparathyroidism. MEN 2 is unusual among cancer syndromes as it is caused by activation of a cellular oncogene, RET. Germline mutations in the gene encoding the RET receptor tyrosine kinase are found in the vast majority of MEN 2 patients and somaticRET mutations are found in a subset of sporadic MTC. Further, there are strong associations ofRET mutation genotype and disease phenotype in MEN 2 which have led to predictions of tissue specific requirements and sensitivities to RET activity. Our ability to identify genetically, with high accuracy, subjects with MEN 2 has revolutionised our ability to diagnose, predict, and manage this disease. In the past few years, studies of RET and its normal ligand and downstream interactions and the signalling pathways it activates have clarified our understanding of the roles played by RET in normal cell survival, proliferation, and differentiation, as well as in disease. Here, we review the current knowledge of the normal functions of RET and the effects of mutations of this gene in tumorigenesis and in normal development.
- multiple endocrine neoplasia type 2
- RET
- receptor tyrosine kinase
Statistics from Altmetric.com
Recognition of cancer as a genetic disease has contributed to the rapid advances of recent years in our ability to identify, diagnose, and treat human neoplasia. Nowhere have the inroads made in these areas been clearer, or had more impact, than in the inherited cancer syndromes, where the presence of multiple, often diverse, disease symptoms and tumour types have made diagnosis and screening highly problematical. The genetic characterisation of many of these diseases in the past few years has provided us with the tools for recognition, screening, and management and has had a huge impact on the perceived burden of these diseases for families. Elucidation of genetic mechanisms and their functional consequences has also given us clues as to the broader systems disrupted in these syndromes which, in turn, have further implications for normal developmental or survival processes. The inherited cancer syndrome multiple endocrine neoplasia type 2 (MEN 2) and its causative gene, RET, are a useful paradigm for both the impact of genetic characterisation on disease management and also for the much broader developmental implications of these genetic events.
The RET receptor tyrosine kinase
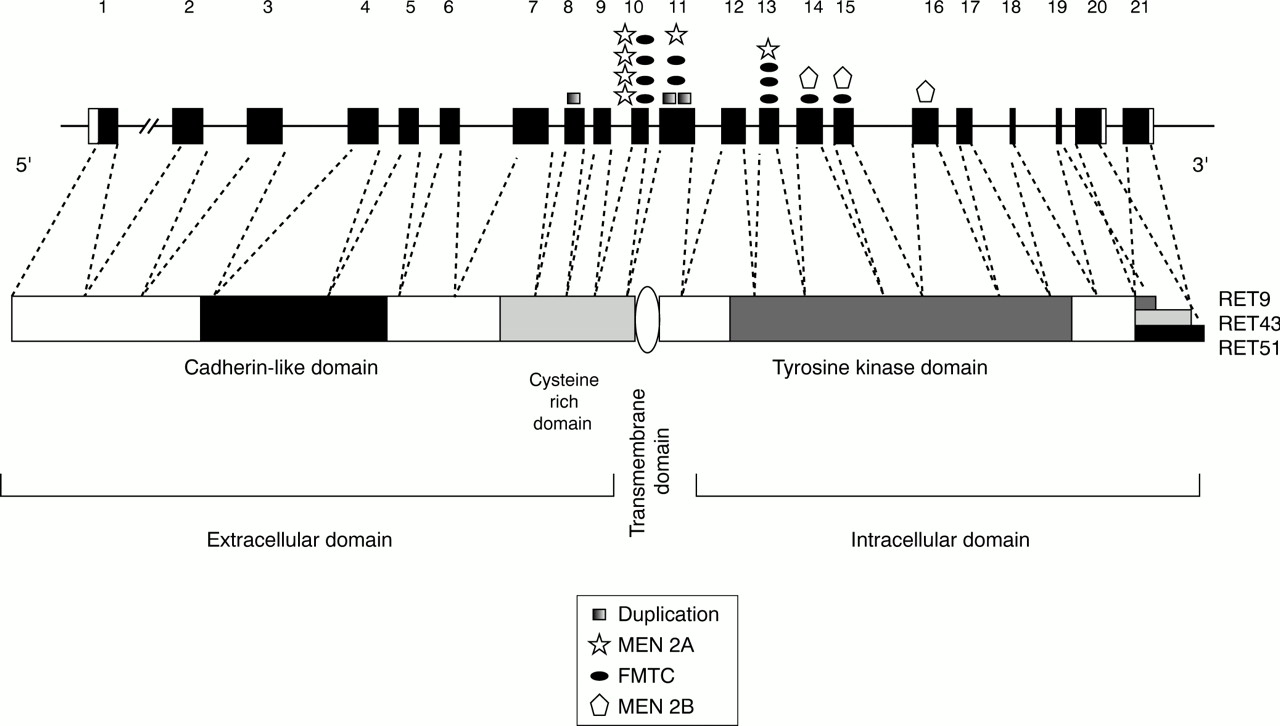
MEN 2 arises as a result of activating mutations of theRET (REarranged during Transfection) proto-oncogene.1-5 RET encodes a receptor tyrosine kinase expressed primarily in neural crest and urogenital precursor cells.6-8 It is a developmentally important gene, required for kidney morphogenesis, maturation of peripheral nervous system lineages, and for differentiation of spermatogonia.6 9 10 Like other receptor tyrosine kinases, the RET protein comprises extracellular, transmembrane, and cytoplasmic domains (fig 1). The extracellular sequences include regions with homology to the cadherin family of cell adhesion molecules and a large cysteine rich region.11-14 Twenty seven of 28 cysteine residues within the cysteine rich domain are conserved between species, suggesting a critical role for these residues in formation of intramolecular disulphide bonds, and thus in determining the tertiary structure of RET proteins.11 13 The intracellular tyrosine kinase domain is similar to that of other receptor tyrosine kinases and functions in phosphorylation of key tyrosine residues involved in interaction with downstream targets and activation of signalling pathways.

Schematic diagram of the structures of the RET gene and protein. Protein domains are indicated. Positions of MEN 2 mutations are shown relative to the 21 RET coding exons and to their corresponding position in the RET protein. Only mutations found in multiple independent families and/or for which functional significance has been confirmed are shown.
Under normal conditions, the RET receptor is activated through a multicomponent complex involving members of two distinct groups of proteins: a soluble ligand of the glial cell line derived neurotrophic factor (GDNF) family, and a cell surface bound coreceptor of the GDNF family receptors α (GFRα) protein family (fig 2). The GDNF proteins are members of the TGF-β subfamily15 and have all been shown to act as potent neuronal survival factors.16 Four members of this family have been identified to date, including GDNF, neurturin (NTN), persephin (PSP), and artemin (ART).16

(A) Schematic diagram showing the members of the RET multimeric signalling complexes and the primary interactions of the different GDNF family ligands and GFRα family members. (B) Predicted model of RET activation by its ligand and coreceptor molecules.18 Soluble ligands bind cell surface bound GFRα family members which, in turn, present the ligands to RET, mediating its dimerisation and autophosphorylation.
Although all four GDNF family members act as the ligands for RET, they do not bind RET directly but first interact with a cell surface bound coreceptor of the GFRα family (fig 2). These coreceptors do not have intracellular domains but are anchored to the cell membrane via a glycosyl-phosphatidylinositol (GPI) linkage.17 18 There are four GFRα family members identified to date (GFRα-1-4).16 GDNF family members form high affinity interactions with a specific member of the GFRα family. GFRα-1 primarily binds GDNF, GFRα-2 binds NTN, GFRα-3 binds ART, and GFRα-4 binds PSP (fig 2).16 All of the different GDNF/GFRα complexes bind to, and activate, RET. The GDNF and GFRα family members have distinct and overlapping expression patterns,16 suggesting that activation of RET by formation of ligand receptor complexes is a tightly regulated process. It is thus not surprising that we see significant disease phenotypes associated with aberrant RET activation.
Multiple endocrine neoplasia type 2 (MEN 2)
MEN 2 is an inherited cancer syndrome characterised by medullary thyroid carcinoma (MTC), a tumour of the neural crest derived parafollicular C cells responsible for the production of calcitonin.19 While MTC, or its precursor lesion C cell hyperplasia, are the most common disease phenotypes in MEN 2 patients (clinically significant disease occurring in 70% of cases), other lesions are also prominent. MEN 2 may be classified into three subtypes based on their occurrence. MTC, phaeochromocytoma (PC), and hyperparathyroidism (HPT) characterise MEN 2A, the most common of these subtypes. PC, a tumour of the adrenal chromaffin cells, occurs in approximately 50% of MEN 2A patients, while only 15-30% of cases develop HPT or parathyroid adenomas.20-22 PC is also present in approximately 50% of those with the MEN 2B subtype. Parathyroid involvement is rare in these cases, and characteristic developmental abnormalities including marfanoid habitus, thickened corneal nerves, and ganglioneuromatosis of the buccal membranes and the gastrointestinal tract are prevalent.19 23 24 MEN 2B is considered to be the most aggressive of the MEN 2 subtypes, with a median age of onset 10 years earlier than seen in MEN 2A.19 24
The third subtype of MEN 2, familial MTC (FMTC), is characterised by the presence of MTC in multiple family members (four or more) as its only disease phenotype.25 Families with a smaller number of MTC cases but without other phenotypes are more difficult to classify as they may represent small FMTC families or MEN 2A families in which PC or HPT have not yet manifested. FMTC is generally considered the least aggressive of the three MEN 2 subtypes with a later onset than MEN 2A or 2B.25
Genetics of MEN 2
MEN 2 is inherited in an autosomal dominant fashion with variable, age dependent penetrance.26 Unlike other cancer syndromes, which are associated with inactivation of tumour suppressor genes, each of the MEN 2 subtypes arises as a result of activating mutations of theRET proto-oncogene. Missense mutations affecting cysteine residues in the RET extracellular domain are found in patients with MEN 2A (fig 1, table 1). Single base pair substitutions in one of five codons, 609, 611, 618, 620 (exon 10) or 634 (exon 11), are found in >98% of MEN 2A families.3 27 28 (fig 1, tables 1 and 2). In each case, these changes result in replacement of a critical cysteine residue by any of several amino acids (table 1). Approximately 87% of MEN 2A mutations affect codon 634 (table 2) and the most frequent substitution at this codon is a cysteine to arginine change (C634R), found in more than 50% of cases.27 28 Rarely, duplication/insertion mutations in exon 11 have been observed in MEN 2A (fig 1), resulting in the insertion of three or four amino acids, including a cysteine residue, within the cysteine rich domain (table 1).29 30As a result of each of the above mutations, a cysteine residue normally involved in the intramolecular disulphide bonds that determine the tertiary structure of RET is unpaired and can form intermolecular bonds with other RET molecules.31-33 The outcome is dimerisation and constitutive activation of the RET tyrosine kinase.31 33
RET mutations and their associated phenotypes1-150
Distribution of RET mutations in MEN 2
Many of the same mutations responsible for MEN 2A have also been found in FMTC (table 1, fig 1). Substitutions of cysteine codons 609, 611, 618, 620, and 634 in exons 10 and 11 are found in more than 80% of FMTC families (table 2).27 28 However, unlike MEN 2A, FMTC mutations are relatively evenly distributed among codons 618, 620, and 634 (table 2).27 28 Less frequently, mutations of cysteine codon 630 and insertions including cysteine residues elsewhere in the cysteine rich region have been identified in FMTC (fig1).34 35 Interestingly, the C634R mutation most common in MEN 2A is not found in FMTC families.27 28 In addition to mutations of extracellular cysteines, FMTC is also associated with amino acid substitutions in the intracellular tyrosine kinase domain. Mutations have been reported in exon 13 (E768D),36-39exon 14 (V804L or V804M),36 40 41 and exon 15 (S891A).42 43 Additional mutations of codons 790, 791, or 844 have been identified in a German patient population but the general distribution of these mutations has not yet been determined.39 The mechanisms by which each of these intracellular mutations activates RET has not been functionally demonstrated; however, they are predicted to alter either ATP or RET substrate binding.36 38 43-45
MEN 2B is associated primarily with a single missense mutation of codon 918 (M918T) which is found in more than 90% of all reported cases (table 2).4 5 27 28 46 Mutations of codon 883 in exon 15 (A883F) have also been shown to occur in a small number of MEN 2B cases.47 48 These variants are exclusively associated with the MEN 2B phenotype. Amino acids 883 and 918 both lie within the substrate binding pocket of the RET tyrosine kinase49 and mutations of these codons result in RET proteins with altered substrate specificity. These altered RET isoforms seem to recognise and phosphorylate substrates preferred by cytoplasmic tyrosine kinases such as c-src and c-abl, rather than the normal substrates of receptor tyrosine kinases.49-51 Recently, MEN 2B associated with a double germline mutation, V804M and Y806C, has been reported.52 This combination of variants has been shown in vitro to be significantly more transforming than either variant alone and this mutant is predicted to have similar biological properties to the other MEN 2B-RET isoforms.53 This double mutation has suggested that multiple low penetrance variants inRET can contribute to a more aggressive disease phenotype.53
Genotype and phenotype
As can be seen from the descriptions above and in table 1, there are strong correlations of MEN 2 disease phenotype and specificRET sequence changes. Mutations affecting cysteine residues in the extracellular cysteine rich domain of RET have been identified in both FMTC and MEN 2A. However, while MEN 2A is associated most frequently with mutations of codon 634, and particularly with the C634R change, mutations in FMTC are more evenly distributed and the C634R mutation is notably absent.27 28 The association ofRET mutations with disease phenotype is even clearer if we consider the occurrence of component tumour types, rather than disease subtypes. Several studies have shown a strong correlation between the presence of a codon 634 mutation and the occurrence of PC and/or HPT in MEN 2 families.3 27 28 54 A further association of the specific C634R mutation with HPT, reported in one large study,3 has been more difficult to confirm on a family as unit basis.21 54 Comparisons between studies have been confounded by differences in screening protocols among studies and, potentially, by patient age and follow up, since HPT is strongly correlated with patient age.21 However, an increased risk of HPT on a person as unit basis in families with C634R mutations has been reported, suggesting that this mutation does confer risk for HPT.21
The genotype/phenotype associations in MEN 2 reflect differences in behaviour and function among the mutant RET isoforms. Protein localisation analyses have shown that RET isoforms with mutations of codons 609, 618, and 620 are not as efficiently translocated to the cell surface as wild type or other mutant RET isoforms.55-58 As described above, these mutations are also associated with lower risks for PC and HPT. Together, these data suggest a model in which there are tissue specific differences in sensitivity to RET activation, with thyroid being most sensitive and parathyroid least sensitive.3 58 According to this prediction, all RET mutations are sufficient to promote tumorigenesis in the thyroid and, consequently, MTC is associated with all MEN 2 subtypes. The adrenal and parathyroid glands are less sensitive to RET activation and, thus, PC and HPT are primarily associated with the most penetrant of RET mutations affecting cysteine 634, where the maximal amounts of mutant RET protein are found on the cell surface.3 58
To date, RET mutations of codons 768, 804, and 891 have been associated primarily with the FMTC phenotype. The risk of other tumour phenotypes is low in subjects with these mutations.27 28 41 The absence of HPT and PC in families with 768, 804, and 891 mutations is consistent with in vitro studies suggesting that RET proteins with these variants have a lower transforming efficiency than isoforms with codon 634 or 918 mutations.44 45 Recent studies have also predicted that V804M mutations particularly may be associated with a later onset and less aggressive disease course.41 59 Together, these data suggest that mutations of RET exons 13, 14, and 15 may represent lower penetrance mutations with a less aggressive associated phenotype, although further families will need to be accrued before this association can be applied clinically.
RET mutations in sporadic tumours
In addition to its role in familial disease,RET mutations have also been implicated in sporadic tumours of MEN 2 type. The majority of MTC cases (>75%) have no associated family history or other indications that might suggest a hereditary cause.19 However, several large population studies have shown that 3-7% of these cases represent occult or de novo MEN 2 cases with germline RETmutations.21 60 61 Approximately 23 to 70% of true sporadic MTC have been shown to harbour somatically occurringRET mutations.62 Analyses of subpopulations of tumour cells or multiple metastases from a single patient have shown that RET mutations are apparent in subsets of clones, suggesting thatRET mutations are not initiating but progressional events in sporadic MTC.63 64 The vast majority of these are the M918T mutation associated with the MEN 2B phenotype, although other mutations of the cysteine rich region and exons 13-15 have been reported. Interestingly, a rare polymorphic sequence variant (c.2439C>T; S836S) was recently shown to be over-represented in patients with sporadic MTC and M918T mutations.65 The significance of this genotype/phenotype association remains to be determined.
Somatic RET mutations are less frequent in PC (10-15% cases),62 perhaps reflecting the lower sensitivity of adrenal chromaffin cells to RET activation.3 58 The M918T mutation is also common here (50%); however, other mutations such as missense mutations of exons 10 and 11 and deletions in the cysteine rich region have been reported.62
RET mutations have not been detected in sporadic HPT or parathyroid adenoma.62 Further, studies of other tumour types including small cell lung carcinoma, neuroblastoma, malignant melanoma, and others have also failed to identifyRET mutations, suggesting that activation of the RET receptor in other cell types may not have significant neoplastic implications.66-68
RET downstream signalling
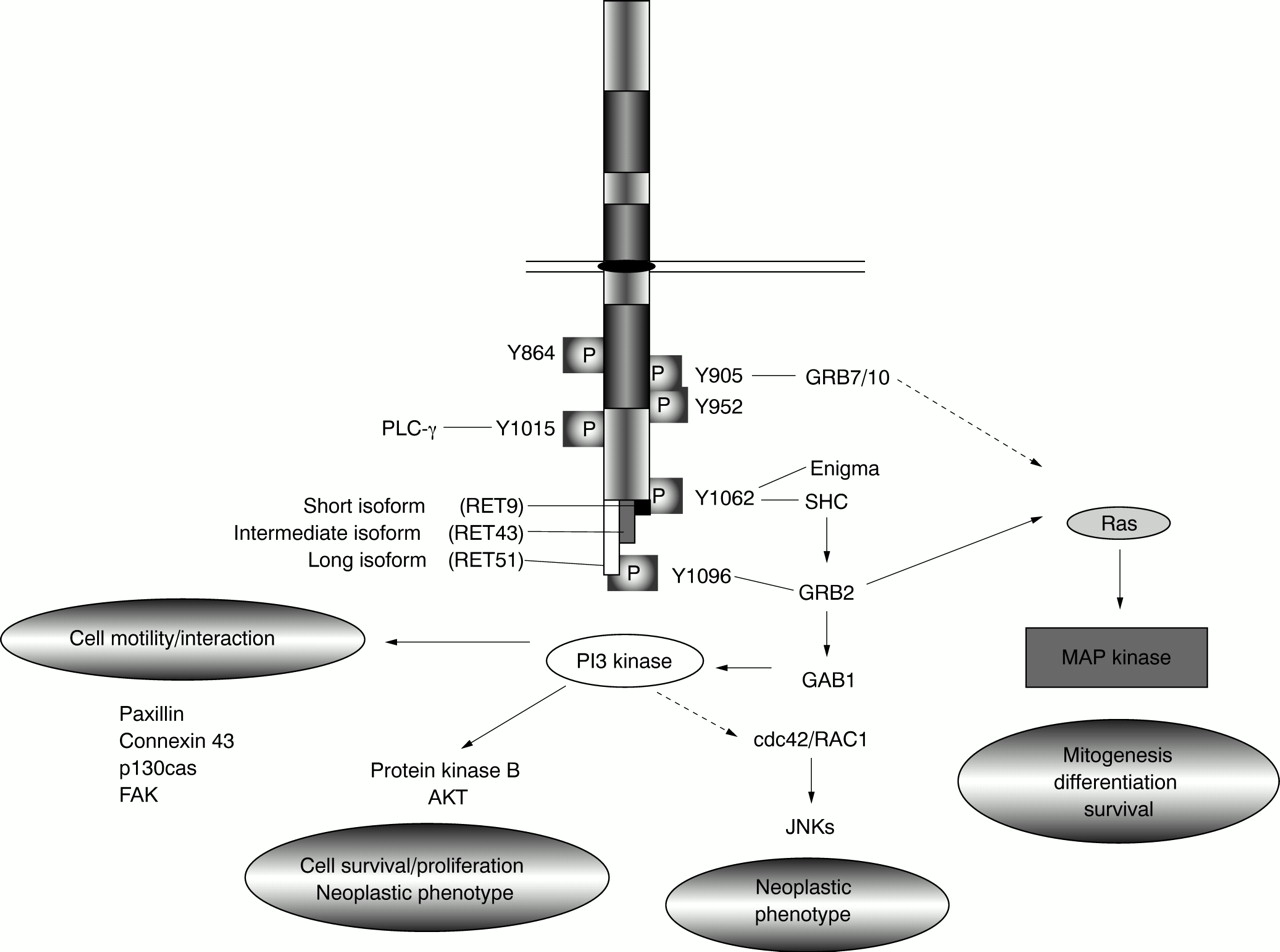
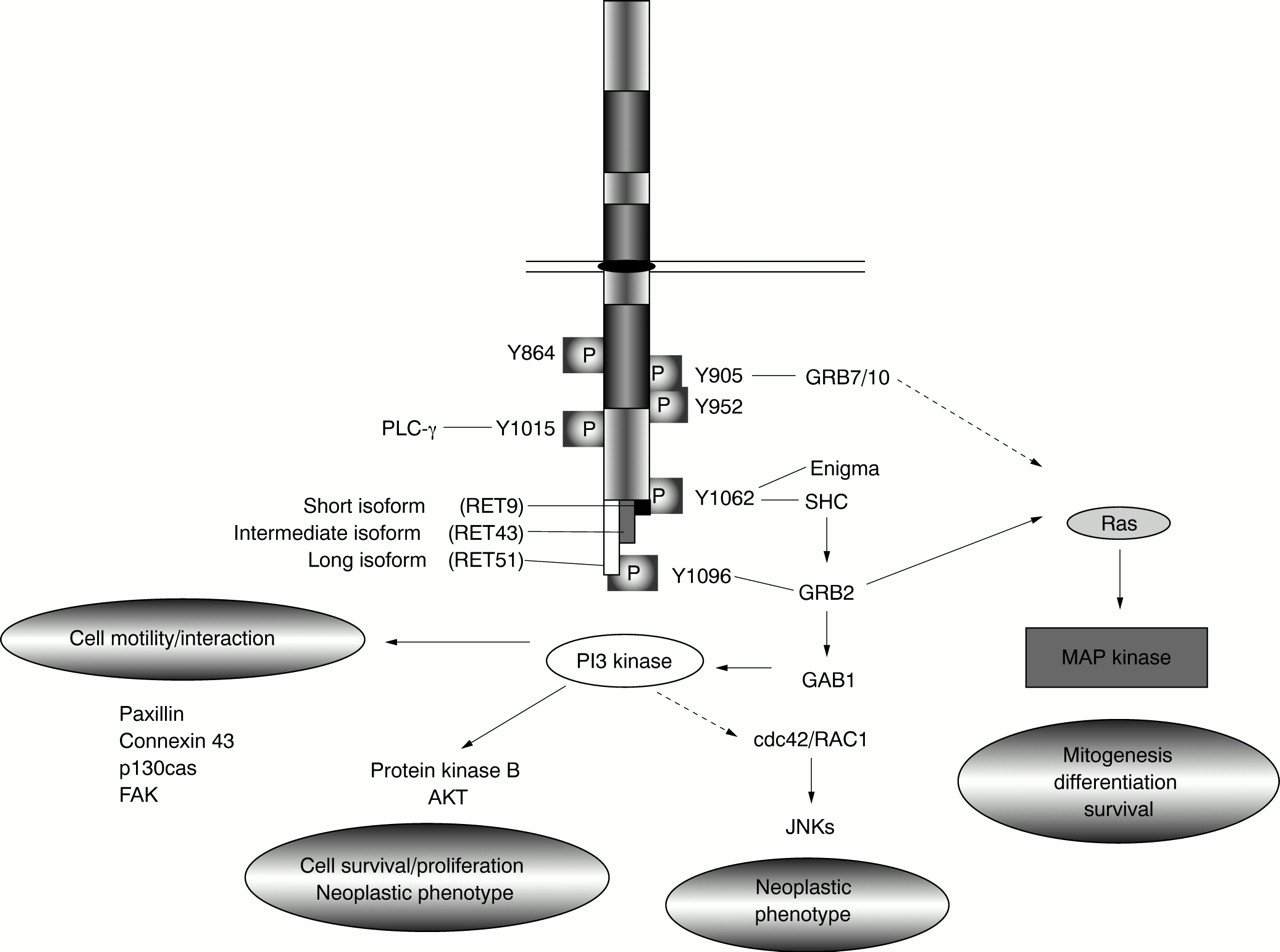
RET's functions in normal and tumorigenic cells are mediated by a complex series of downstream interactions. RET can trigger growth, differentiation, or survival responses, depending on the cell type or developmental stage in which it is activated. Dimerisation of RET receptors, either owing to the presence of activating mutations or ligand binding, results in autophosphorylation of intracellular tyrosine residues. To date, nine of RET's 18 intracellular tyrosines have been shown to be phosphorylated upon RET activation.11 69 Adaptor proteins containing SH2 domains recognise the amino acid sequences surrounding these phosphorylated residues and target these sites specifically. Interactions identified to date include GRB7 and 10, binding at tyrosine 905 (Y905), phospholipase C-γ (PLC-γ) at Y1015, SHC at Y1062, and GRB2 at Y1096 (fig 3).31 70-76 RET also interacts with enigma, a cytoplasmic, membrane anchored LIM domain protein which binds RET Y1062 in a phosphorylation independent manner and may play a role in positioning the RET tyrosine kinase with respect to the cell membrane.76
{kind=link}
{kind=link}
{kind=link}
Schematic diagram showing some of the downstream pathways activated by RET. Specific downstream interactions confirmed in vitro are indicated. Dotted lines indicate predicted signalling paths not yet confirmed in this system.
RET activates a number of well characterised downstream signalling pathways through interactions with these adaptor proteins (fig 3). RET has been shown to stimulate RAS mediated activation of the MAP kinase pathway which is required for both neuronal survival and differentiation.77-81 The RAS-MAP kinase cascade is activated through SHC and GRB2 (Y1062) or directly through GRB2 binding (Y1096). GRB7 or 10 binding (Y905) has also been linked to activation of this pathway.82 In some cell types, RET mediated activation of RAS dependent pathways can result in cellular differentiation,81 while in other cell types, RET activation may actually block RAS induced differentiation by preventing nuclear translocation of ERK and induction of immediate early gene transcription.83
Activation of phospho-inositol 3 (PI3) kinase, probably through RET's interaction with GRB2 and GAB1 and recruitment of the PI3 kinase regulatory subunit, has also been implicated in cell proliferation and survival.51 84 RET stimulation of this pathway is further associated with cell motility and cellular interactions and can lead to lamellapodia formation and phosphorylation of proteins linked to formation of focal adhesions (for example, paxillin, focal adhesion kinase (FAK), Crk associated substrate p130cas) and to increased expression of the gap junctional protein connexin 43 (fig3).50 80 85-89 RET also acts through PI3 kinase to stimulate protein kinase B/AKT activation which plays a role in cell survival and proliferation but also contributes to RET's oncogenic potential.51 84 88 RET has also been shown to activate the c-jun N-terminal kinase (JNK) pathway through a cdc42/RAC1 small GTPase which contributes to establishment of the neoplastic phenotype.90 It is not yet clear how this is triggered by RET, but JNKs have been shown to lie downstream of PI3 kinase in other systems.
Finally, RET has been shown to interact with PLC-γ through Y1015. This association is also required for full oncogenic activation of RET.71 91
As MEN2A and2B-RET mutations contribute to RET activation via different mechanisms, it is perhaps not surprising that different tyrosine residues appear to be significant to transformation in each case. Phosphorylation of tyrosines 864 and 952 is crucial to transformation associated with the MEN 2B mutation of codon 918, while Y905 phosphorylation is essential to transformation in the context of MEN 2A mutations.69 82 Other tyrosines, such as Y1015, Y1062, and Y1096, are phosphorylated in all mutation contexts and are universally required for transformation,73 92-94although the extent of activity may vary. For example, in response to MEN 2B type RET mutations, Y1096 has relatively reduced phosphorylation while RET stimulation of PI3 kinase is relatively increased, as compared toMEN2A mutations.50 51 69 90
The downstream interactions of RET are further complicated by alternative splicing of 3′ exons which results in three protein isoforms with nine (RET9), 51 (RET51), or 43 (RET43) unique C-terminal amino acids (figs 1 and 3).11 95 96 These isoforms differ in their relative binding of adaptor proteins to Y1062 which lies in three distinct amino acid contexts in these proteins.75 97 98 Further, Y1096, discussed above, is present in only the RET51 protein isoform. Consistent with these differences, RET isoforms have different efficiencies in inducing transformation or differentiation in vitro and distinct developmental expression patterns in vivo.75 97-99
Diagnosis and management of MEN 2
Recognition of the genetic events responsible for MEN 2 have significantly improved our ability to diagnose and manage the disease. The prognosis for MEN 2 patients is very good with early diagnosis and intervention. Previously, repeated biochemical testing of at risk subjects was required to identify those manifesting MEN 2 symptoms. Blood pressure monitoring and quantitation of 24 hour urine catecholamines were used to identify adrenal hyperplasia or PC.100 A provocation assay for increased calcitonin or calcium release (indicative of C cell hyperplasia) was used to screen subjects at risk for MTC.100 A raised test result indicated the presence of C cell hyperplasia or MTC which would be managed surgically. Unfortunately, these tests were subject to false positive results and were notoriously inaccurate in young children, where testing was most critical.101 102 However, the major disadvantage of these tests was that they did not distinguish the family member who had not inherited a MEN2mutation, and was therefore not at risk of the disease, from presently asymptomatic subjects who would in future develop the disease phenotype.
Currently, early genetic screening for RETmutations is considered the standard of care for MEN 2.100 103 The advent of genetic testing methods has made it possible to detect those people who have inherited aMEN2-RET mutation before the onset of disease symptoms and the associated morbidity or mortality. Predictive and diagnostic testing is facilitated by the high incidence of detectable mutations (>95%) and the small number of target codons.27 28 If a member of a family with a knownMEN2-RET mutation is found not to have that mutation, they are not considered at risk of MEN 2 and should not require further screening. In the case of subjects where theMEN2-RET mutation is found, prophylactic thyroidectomy is recommended before the age of 6 years,103since MTC has been diagnosed at very early ages in MEN 2 families.104 In cases of MEN 2B, which is associated with an earlier age of onset and more aggressive disease than other MEN 2 subtypes, surgery before the age of 3 may be warranted.100 101 103 104
Although current genetic tests can identify the vast majority (>98%) of MEN 2 cases,27 28 there remain a few families for which a RET mutation has not been identified and subjects for whom it has not been possible to exclude a familial origin for MTC. In these cases, family members must be treated as in the pre-DNA testing era with frequent biochemical screening of at risk subjects.
Other RET related phenotypes
In addition to MEN 2, RET has been implicated in several other pathologies including papillary thyroid carcinoma and Hirschsprung disease.
PAPILLARY THYROID CARCINOMA
Papillary thyroid carcinoma (PTC) is a tumour of the thyroid follicular cells which is associated with frequent somatically occurring rearrangements of the RETgene.105 106 As a result of either chromosomal translocation or inversion, the 3′ portion of theRET gene encoding the intracellular domains becomes juxtaposed to 5′ sequences from one of several other genes (H4, RIα, ELE1, HTIF1, GOLGA5, RFG7). Although these partner genes have diverse normal functions, each appears to contribute a domain with dimerisation potential to the RET fusion protein.105 106 The resultant chimeric molecule can dimerise and autophosphorylate constitutively to activate downstream signalling in a cell type in whichRET is not normally expressed, leading to cellular transformation. Recent studies have shown that the incidence of RET mutations in PTC is significantly higher in people who have suffered radiation exposure, as in the Chernobyl region.106 Initially, rearrangements juxtaposing the ELE1 and RETgenes are the most common in those subjected to higher radiation doses, suggesting that this chimeric protein may contribute to rapid tumour formation.106 However, at longer intervals after exposure, the frequency of RET rearrangements has declined and a shift towards H4/RET chimeras has been noted.106
HIRSCHSPRUNG DISEASE
Hirschsprung disease (HSCR) is a developmental disorder marked by the lack of innervation of variable lengths of the hind gut.107 HSCR affects approximately 1/5000 neonates and may occur in either sporadic or heritable forms.107Inactivating mutations of RET, including deletions, insertions, and point mutations, have been detected in a subset of HSCR patients.62 108 The outcome of these changes is to reduce the amount of functional RET protein on the cell surface, resulting in haploinsufficiency for RET.45 55 56 109 110 GermlineRET mutations are found in approximately 40% of familial HSCR cases and in 3-7% of sporadic HSCR cases.111 112 Several studies have also identified mutations of the RET ligands GDNF and NTN in patients with HSCR,113-116 although mutations of the GFRα family are surprisingly absent.117-119 These mutations are thought to act primarily as modifiers of the RETmutation phenotype in HSCR. Recent studies have also shown an association between the HSCR phenotype and polymorphisms in exons 2, 13, and 15 of RET in sporadic HSCR, even in the absence of RET mutations, suggesting that the level of RET function, even within the normal range, has implications for expression of the HSCR phenotype.120 121The co-occurrence of mutations or variants ofRET, mutations at other loci such as the endothelin B receptor gene,122 123 and an association with an as yet unknown locus on chromosome 9q31124 support a multigenic origin for HSCR in which RET is only one of the players, albeit a significant one.
In rare families, HSCR and MEN 2A or FMTC may co-occur, because of the same RET mutation.108 125 126These phenotypes are generally associated with cysteine to arginine changes in exon 10 of RET. As described above, protein isoforms containing these specific mutations have been shown to translocate to the cell surface with low efficiency.55 57 58 As a result, insufficient RET protein is available in the developing enteric nervous system and HSCR can result, while at the same time activation of the RET signalling pathways in the thyroid/adrenal glands is sufficient to permit hyperplasia or tumour formation.
RET and other pathologies: from cause to cure?
To date, RET has not been implicated in other disease pathologies, but it remains of interest as a target of therapies in patients with neurodegenerative disease or nerve injuries. The GDNF family of RET ligands are well known for their neuroprotective properties, especially in dopaminergic neurones.16 In particular, activation of RET by GDNF ligands promotes survival of dopaminergic neurones and has thus been of interest as a target for therapies in diseases such as Parkinson's disease.127-129 A variety of studies have also shown that signalling through RET is increased in response to a range of insults and may represent an important survival factor for neurones exposed to physical or chemical insult. The clinical implications of these findings have yet to be explored.
Recent studies have shown that RET is required for normal development of spermatogonia and, hence, normal sperm formation10 and a potential role for RET in therapies for male infertility has been proposed.
Conclusion
It has been less than 10 years since the relationship betweenRET mutations and MEN 2 was first described. In that time, we have revolutionised the management and prognosis for this disease in ways rarely possible for human cancers. The study of RET, and its role in normal development, have since then expanded in new directions and identified other disease associations and yet our knowledge of the system is still very preliminary. In the next few years we can predict that our understanding of RET will turn towards its normal functions and how we may use these in treatment or mitigation of diseases for which we are only beginning to predict a role for RET.