Article Text

Abstract
Papillon-Lefèvre syndrome (PLS) is an autosomal recessive disorder characterised by palmoplantar hyperkeratosis and severe early onset periodontitis that results in the premature loss of the primary and secondary dentitions. A major gene locus for PLS has been mapped to a 2.8 cM interval on chromosome 11q14. Correlation of physical and genetic maps of this interval indicate it includes at least 40 ESTs and six known genes including the lysosomal protease cathepsin C gene (CTSC). The CTSCmessage is expressed at high levels in a variety of immune cells including polymorphonuclear leucocytes, macrophages, and their precursors. By RT-PCR, we found CTSC is also expressed in epithelial regions commonly affected by PLS, including the palms, soles, knees, and oral keratinised gingiva. The 4.7 kbCTSC gene consists of two exons. Sequence analysis of CTSC from subjects affected with PLS from five consanguineous Turkish families identified four different mutations. An exon 1 nonsense mutation (856C→T) introduces a premature stop codon at amino acid 286. Three exon 2 mutations were identified, including a single nucleotide deletion (2692delA) of codon 349 introducing a frameshift and premature termination codon, a 2 bp deletion (2673-2674delCT) that results in introduction of a stop codon at amino acid 343, and a G→A substitution in codon 429 (2931G→A) introducing a premature termination codon. All PLS patients were homozygous for cathepsin C mutations inherited from a common ancestor. Parents and sibs heterozygous for cathepsin C mutations do not show either the palmoplantar hyperkeratosis or severe early onset periodontitis characteristic of PLS. A more complete understanding of the functional physiology of cathepsin C carries significant implications for understanding normal and abnormal skin development and periodontal disease susceptibility.
- cathepsin C
- palmoplantar hyperkeratosis
- Papillon-Lefèvre syndrome
- periodontitis
Statistics from Altmetric.com
Papillon-Lefèvre syndrome (PLS) is an autosomal recessive palmoplantar keratodermal disorder.1 In the recently proposed nosology for the palmoplantar keratodermas, PLS is grouped with the palmoplantar ectodermal dysplasias.2 Clinically, PLS (MIM 2450003) is characterised by hyperkeratosis of the palms and soles and severe early onset periodontitis that results in the premature loss of both the primary and secondary dentitions (fig1). The frequency of the disorder is approximately 1-4/million people, with parental consanguinity reported in almost half of PLS cases.4 Clinically, it is the unique association of severe periodontitis that distinguishes PLS from other palmoplantar keratodermas. The periodontal disease associated with PLS is particularly aggressive and unresponsive to traditional periodontal therapies. As a result, most patients become edentulous by 20 years of age. In addition to the cardinal features of PLS, some PLS patients are reported to have an increased susceptibility to infections.5

Clinical photographs showing palmoplantar keratosis and periodontal disease in PLS study patient. (a) Palmar hyperkeratotic lesions. (b) Plantar hyperkeratotic lesions. (c) Hyperkeratotic lesions affecting the knees. (d) Periodontitis involving erupting permanent dentition. (e) Periapical radiographs showing severe alveolar bone loss affecting erupting permanent teeth.
Three groups have independently used homozygosity linkage mapping strategies to localise a major locus for PLS to overlapping genetic intervals on chromosome 11q14.6-8 Integration of the different genetic maps used in these linkage studies placed thePLS locus within a 2.8 cM interval flanked by D11S4197 and D11S931.9 Correlation of this integrated genetic map with physical maps of the interval suggests it contains at least 40 ESTs and six known genes. Nine of these ESTs and genes are normally expressed in oral keratinised gingival tissues. As part of our effort to identify the gene mutation(s) responsible for PLS, we performed mutational analysis for those genes localised to thePLS candidate interval that were also expressed in gingiva.9 In this report, we describe four different mutations of the cathepsin C gene that were identified in subjects affected with PLS from five consanguineous Turkish families.
Methods
FAMILY MATERIAL AND CLINICAL DIAGNOSIS
Five Turkish families have been described previously.8 All available family members provided consent for the study and were clinically examined. A diagnosis of PLS was made in subjects with severe early onset periodontitis and the clinical appearance of hyperkeratosis on the palmar and plantar surfaces. All affected subjects also had hyperkeratosis on the knees. DNA was isolated from peripheral blood samples from all available members of these nuclear families using standard techniques (Qiamp Blood Kit, Qiagen).
RNA ISOLATION, AMPLIFICATION, AND TISSUE EXPRESSION ANALYSIS.
Total RNA was generated from fresh tissue samples (gingiva, palm, sole, knee) using TRIZOL reagent (Molecular Research Center Inc, Cincinnati, OH) according to the manufacturer's protocol. To determine if cathepsin C was expressed in a given tissue, single tube RT-PCR was carried out using the Access RT-PCR System (Promega, Madison, WI), following the manufacturer's protocol. A portion of each reaction was visualised following agarose gel electrophoresis in the presence of ethidium bromide. Amplification primers located within exon 1 F 5′-AGGAGGTTGTGTCTTGTAGCC-3′ (nt 2127-2147) and exon 2 R 5′-AGTGCC TGTGTAGGGGAAGC-3′ (nt 3896-3877) produce an amplicon of 123 bp from cDNA. A standard PCR protocol was followed with an annealing temperature of 65°C.
GenBank accession numbers are full length cDNA ofCTSC (NM-001814) and full length genomic DNA of CTSC (U79415).
SEQUENCING AND MUTATION ANALYSIS
PCR primers were designed to cover the entire cathepsin C gene in overlapping fragments, from 1075 nucleotides 5′ to the start codon (nt1271) to 240 nucleotides 3′ to the termination codon (nt4307) using cathepsin C (DPP-I) sequence data (Accession No U79415) (table 1). The PCR products were prepared for sequencing by excising the bands from the agarose gel and extracting the fragments using a Qiagen Gel Clean-up Kit. The sense and antisense strand of each PCR product were directly sequenced on an ABI Prism 310 Genetic Analyzer (Perkin-Elmer) using four dye terminator chemistry. Approximately 1-3 ng of purified product and 3.2 pmol primer were added to premixed reagents from the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit, FS (Perkin-Elmer) and underwent a cycle sequencing reaction in a GeneAmp PCR System 9700 (Perkin Elmer). The linear amplification started with a 10 second denaturation at 96°C, five seconds annealing at 50°C, and four minutes extension at 60°C. The fluorescently labelled sequencing products were separated from residual reaction reagents using a Centri-Sep spin column (Princeton Separations, Aldelphia, NJ) and electrophoresed on POP6 capillary at 1500 V for 30 minutes. Sequencing data were automatically collected and analysed by the ABI Prism 310 software.
PCR primers
We analysed raw sequence data, generated consensus sequences, and produced nucleotide/amino acid alignments (DNASIS V2.6 for Windows, Hitachi Software Engineering Co Ltd). Mutations were detected by creating nucleotide/amino acid alignments of reported cathepsin C sequence data versus affected PLS patients' sequence data using the Higgins-Sharpe UPGMA.
Results
PLS FAMILIES
Parents of all families were consanguineous (uncle-niece marriage). Linkage studies localised a PLS gene in these five families to chromosome 11q14.8 Affected subjects were each homozygous for SSTR markers within the PLS candidate interval on chromosome 11q14, consistent with inheritance of both maternal and paternal copies of this genetic interval from a common familial ancestor (“identical by descent”). Four different haplotypes for SSTR markers spanning the critical region were identified (fig 2), consistent with four independent mutations in the gene responsible for PLS.

Haplotype data for chromosome 11q SSTR markers spanning the PLS gene locus. Segments which are likely to be homozygous by descent are boxed. Arrows indicate recombinant events. Subjects 7 and 22 share a common haplotype for D11S1979, D11S1887, D11S1780, D11S1367, D11S931, and D11S4175.
ANALYSIS OF CATHEPSIN C
Using RT-PCR we found that cathepsin C is normally expressed in epithelium from palms, soles, knees, and keritinised oral gingiva from unaffected subjects (data not shown). The cathepsin C gene spans approximately 4.7 kb and consists of two exons. Sequence analysis of exonic, intronic, and the 5′ regulatory regions of the cathepsin C gene, CTSC, showed that affected subjects from these PLS families were homozygous for four different mutations that significantly altered the cathepsin C open reading frame.
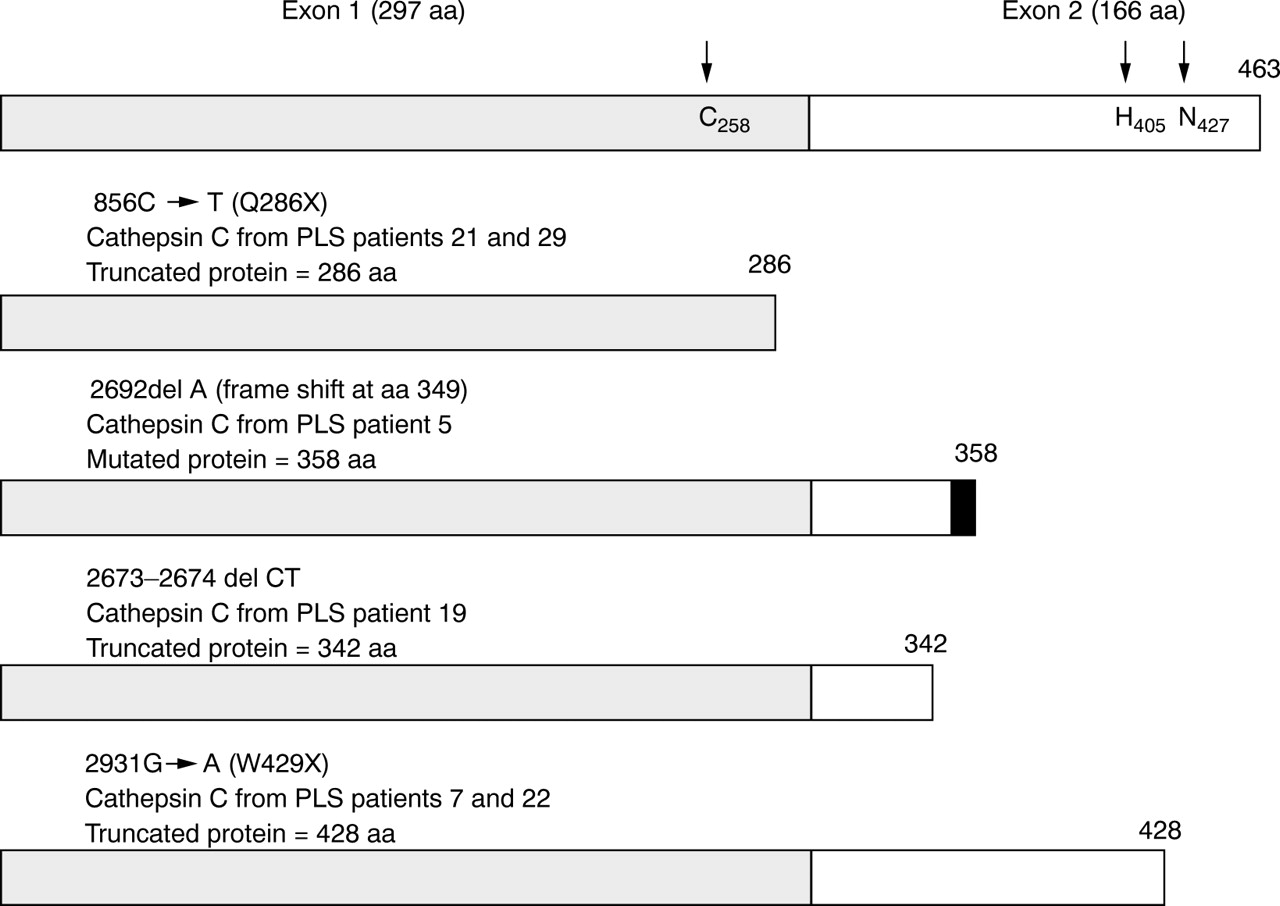
Two affected subjects from one family were found to have an exon 1 nonsense mutation (856C→T), which introduces a stop codon at amino acid 286 (fig 3). Three different exon 2 mutations were detected (fig4). A deletion of a single nucleotide (2692delA) of codon 349 was found that introduced a frameshift and a premature termination codon (TGA) 27 bases downstream of the mutation. This mutation is predicted to result in a protein of 358 amino acids, compared to the normal (wild type) 463 amino acids. A deletion of two bases of codon 343 (2673-2674delCT) resulting in the introduction of an early termination codon (TGA), which is predicted to result in a truncated protein of 342 amino acids, was identified in another family. A G→A substitution in codon 429 (2931G→A) that altered the original TRP codon (TGG) to a terminator codon (TAG), W429X, was identified in two affected subjects (Nos 7 and 22) from two additional families. The expected truncated protein is 428 amino acids. Although these families were not known to be related, the fact that affected subjects from these two Turkish families are homozygous for a common cathepsin C gene mutation and also share a common haplotype for SSTR markers in the PLS candidate interval flanking the cathepsin C gene (D11S1979-D11S4175) suggests that they have inherited the same cathepsin C gene mutation from a common ancestor. Summaries of the mutations and the predicted protein products are shown in fig 5.

Pedigree and sequence analysis of CTSC exon 1. The numbering of the wild type sequence shown above the figure is based upon the genomic sequence of CTSC (Accession U79415). Filled symbols indicate affected subjects. Half shading indicates carriers based upon DNA sequencing results. All affected subjects are homozygous. Arrows indicate the position of the mutation. This family has a nonsense mutation (856 C→T) at codon 286 resulting in a truncated protein of 286 amino acids.

Pedigrees and sequence analysis of CTSC exon 2 for four families with PLS. Symbols are as described for fig 3. (A) Family with a single base pair deletion (2692delA) of CTSC resulting in a frameshift and premature termination. (B) Family with a 2 bp deletion (2673-2674delCT) of CTSC resulting in a frameshift and premature termination. (C, D) Families with a nonsense mutation (2931G→A) at codon 429 resulting in a truncated protein of 428 amino acids. The father in family C is dead and no sample was available for analysis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of predicted consequences for truncated cathepsin C molecules. The wild type cathepsin C with 463 amino acids is shown at the top, as predicted from the coding region of cloned CTSC NCBI Accession No U79415. The three residues of the active site are indicated by arrows. The stippled box represents consensus exon 1 coded region; the white box indicates consensus exon 2 coded region; the black box indicates altered translational frame.
Discussion
Papillon-Lefèvre syndrome is a palmoplantar keratoderma (PPK) with the characteristic clinical features of palmoplantar hyperkeratosis and severe periodontal destruction. The PPKs are a heterogeneous group of diseases all having gross thickening of the palmoplantar skin. Clinically, the finding that distinguishes PLS from other PPKs is severe, early onset periodontal destruction. In affected subjects, the development and eruption of the primary teeth proceed normally, but the eruption of these teeth into the oral cavity is associated with gingival inflammation and subsequent rapid destruction of the periodontium. This form of destructive periodontitis is characteristically unresponsive to traditional periodontal treatment modalities and, consequently, the primary dentition is usually exfoliated prematurely. After exfoliation, the inflammation subsides and the gingiva resumes a healthy appearance. However, with the eruption of the permanent dentition the process is usually repeated, resulting in the premature exfoliation of the permanent dentition, although the third molars are sometimes spared.5Destruction of the alveolar bone in PLS is usually severe, resulting in generalised atrophy of the alveolar ridges, further complicating dental therapy.
Because the gene encoding cathepsin C was localised to the refined PLS candidate interval on chromosome 11q14 and was normally expressed in epithelium from sites affected by PLS, it was evaluated as a candidate gene for PLS. Cathepsin C, or dipeptidyl aminopeptidase I (EC3.4.12.1), is a lysosomal cysteine protease capable of removing dipeptides from the terminus of protein substrates, but at higher pH it also exhibits dipeptidyl transferase activity.10 The cathepsin C gene spans approximately 4.7 kb and consists of two exons that encode a 463 amino acid polypeptide with predicted features of the papain family of cysteine proteases.11 Unlike cathepsin B, H, L, and S, which are small monomeric enzymes, cathepsin C is a large (200 kDa) oligomeric protein that consists of four identical subunits, each composed of three different polypeptide chains.12 13Expression of CTSC is tissue dependent.14 CTSC is expressed at high levels in lung, kidney, placenta, and cells of myeloid origin, and at moderate or low levels in a variety of other organs.15 The CTSC message is expressed at high levels in immune cells including polymorphonuclear leucocytes and alveolar macrophages and is also expressed at high levels in osteoclasts.11 16 The pathological clinical findings of the PLS patients studied here involve severe inflammation and destruction of the gingiva as well as hyperkeratosis of the skin from palmar, plantar, and knee sites. In unaffected subjects, cathepsin C is normally expressed in epithelial tissues from sites clinically affected by PLS.
The parents of the PLS patients in this study are consanguineous. As a result, the patients in each family are homozygous for cathepsin C mutations inherited from a common ancestor. As parents and several sibs who are heterozygous carriers for CTSCmutations do not appear to show either the palmoplantar hyperkeratosis or severe early onset periodontitis characteristic of PLS, it appears that the presence of one wild type CTSC is sufficient to prevent PPK and periodontal destruction. A consistent finding in the three linkage reports to date is the lack of a common haplotype among affected subjects from different families. The present report describes four different germline homozygous CTSC gene mutations associated with PLS. These findings suggest that theCTSC mutations responsible for PLS have probably arisen independently. All mutations reported here result in the introduction of premature stop codons. While the W429X mutation encodes a protein shortened by only 35 amino acids, the introduced stop codon is one amino acid from the asparagine residue in the active site (fig 5). It is likely that such a mutation would cause a conformational alteration that may decrease or abolish activity. Additionally, we have also identified single nucleotide changes that result in missense amino acid changes in several additional PLS patients from other populations, suggesting that structural alterations of cathepsin C may cause PLS.
In addition to the cardinal features of PLS, reports suggest some PLS patients have an increased susceptibility to infections.5This generalised increased susceptibility to infection may reflect the more deleterious effects of specific cathepsin C mutations, or may reflect the epigenetic effects of other gene loci. An increased susceptibility to infections was not reported for the affected subjects reported here. A variety of immunological findings have been reported in PLS patients, including decreased monocyte chemotaxis, decreased neutrophil chemotaxis, impaired neutrophil phagocytosis, altered superoxide production, and decreased blastogenic response, but it has been difficult to extrapolate results of these studies. Consequently, the underlying pathogenesis of PLS has been poorly understood.17 Immunological findings previously reported for affected subjects from the current families includes decreased PMN chemotaxis and increased CD11b expression.18 19 The pathological clinical findings associated with PLS suggest that cathepsin C is functionally important in the structural growth and development of skin and in susceptibility to periodontal disease. As a lysosomal cysteine proteinase, cathepsin C is important in intracellular degradation of proteins and appears to be a central coordinator for activation of many serine proteinases in immune/inflammatory cells.11 It is unknown if the profound periodontal disease susceptibility is a consequence of altered integrity of junctional epithelium surrounding the teeth. It is interesting that once teeth are exfoliated, and consequently the junctional epithelium is eliminated, the severe gingival inflammation resolves. A more complete understanding of the functional physiology of cathepsin C carries significant implications for understanding periodontal disease susceptibility. Identification of cathepsin C gene mutations in PLS raises the possibility of creating an animal model to study the development, treatment, and prevention of hyperkeratosis and periodontitis.
Classification of the PPKs based upon histological findings, epidermolysis, and localisation of lesions within the skin (diffuse, linear, or focal) has not been helpful in understanding the pathomechanism of disease.20 Identification of mutations in specific genes has led to development of a revised nosology of these diseases in which PLS is grouped with the palmoplantar ectodermal dysplasias.2 In addition to providing insight into both normal and abnormal epithelial growth and development, identification of mutations in cathepsin C associated with PLS will contribute to the overall nosology of the PPKs. For example, the differential diagnosis of PLS includes Haim-Munk syndrome (MIM 245010), which, in addition to PPK and severe periodontal destruction, is clinically characterised by pes planus, recurrent pyogenic skin infections, and possibly arachnodactyly.21 22 Gorlin et al 22 23 have suggested that Haim-Munk syndrome (HMS) may be an allelic variant of PLS. Identification of cathepsin C mutations in PLS affected subjects now permits studies to determine if HMS and PLS are allelic variants.
Acknowledgments
We thank the families and Dr Mark Pettenati for participation in these studies supported by NIDCR grants DE11601 and DE12920. We also thank Tracie Stivason and Ruby Griffin for help with manuscript preparation.