Article Text

Abstract
Two unrelated mildly retarded males with inversions of the X chromosome and non-specific mental retardation (MRX) are described. Case 1 has a pericentric inversion 46,Y,inv(X)(p11.1q13.1) and case 2 a paracentric inversion 46,Y,inv(X) (q13.1q28). Both male patients have severe learning difficulties. The same chromosomal abnormalities were found in their mothers who are intellectually normal. Fluorescence in situ hybridisation mapping showed a common area of breakage of each of the inverted chromosomes in Xq13.1 near DXS131 and DXS162. A detailed long range restriction map of the breakpoint region was constructed using YAC, PAC, and cosmid clones. We show that the two inverted chromosomes break within a short 250 kb region. Moreover, a group of ESTs corresponding to an as yet uncharacterised gene was mapped to the same critical interval. We hypothesise that the common inversion breakpoint region of the two cases in Xq13.1 may contain a new MRX gene.
- inverted X chromosome
- non-specific X linked mental retardation
- XLMR
- MRX
Statistics from Altmetric.com
X linked mental retardation (XLMR) represents a large group of disorders where mental retardation (MR, IQ<70) is a feature of the phenotype and the corresponding gene is on the X chromosome (lod score >2.0). Formally, the XLMR group of disorders is subdivided into two major groups1: syndromal XLMR where MR is an accompanying feature of a usually more complex, dysmorphic phenotype, and non-specific XLMR or MRX where MR is the only common feature of the phenotype. While the identification of numerous genes for syndromal XLMR can take advantage of the analysis of several families with that particular syndrome, the situation with MRX is complex mainly because of the genetic heterogeneity underlying an otherwise clinically homogeneous phenotype. To date more than 105 syndromal XLMR disorders and more than 65 MRX disorders have been accumulated world wide.2 The prevalence of MRX based on a study from British Columbia was calculated to be 1.83 per 1000 males.3However, the true prevalence remains uncertain owing to the mild nature of the phenotype segregating in some families.
To date, four MRX genes have been identified:FMR2 as the FRAXE fragile site associated MRX gene4 ,5; the oligophrenin-1 gene interrupted by an X;12 translocation and mutated in the MRX60 family6; theGDI1 gene mutated in families MRX48, MRX41,7 and family R8; and thePAK3 kinase with mutation detected in family MRX30.9 Three of these genes appear to be involved in molecular signalling pathways involving small GTPases (GDI and oligophrenin-1)10 and kinase cascades (PAK3).9
Altogether, there have been a minimum of eight MRX genes predicted3 ,11 based on non-overlapping linkage intervals. Owing to the genetic heterogeneity and often large (several cM) linkage intervals, the positional cloning or positional candidate approaches can be difficult to carry out. A suitable, if not the only, alternative is to identify candidate MRX genes from X chromosomal rearrangements (translocations, inversions, deletions) associated with an MR phenotype and to test these candidate genes in MRX families mapping across the rearranged region. Only a few have been described so far,2 ,12-18 but they constitute a highly valuable resource since two out of four familial MRX genes were cloned using such a rearrangement.
We have identified two inverted X chromosomes, one with a pericentric inversion 46,Y,inv(X)(p11.1q13.1) and the second with a paracentric inversion 46,Y,inv(X)(q13.1q28) associated with non-specific mental retardation in both cases. We present here the fine physical mapping of two inversion breakpoints in the Xq13.1 region near DXS131 and DXS162. In addition, a group of ESTs represented by EST R97207 has been mapped to the critical interval.
Methods
FLUORESCENCE IN SITU HYBRIDISATION
Initial cytogenetic analysis indicated that one of the inversion breakpoints from each patient maps to the Xq13 region. Individual YAC clones either from established19 or unpublished YAC contigs (Villard et al, unpublished data) were used to narrow down the interval of the two breakpoints. Once the YACs crossing the breakpoints were identified, cosmid clones were screened from available libraries (LLNL X chromosome cosmid library provided by the HGMP Resource Center).
The probes (DNA from YACs or cosmids) for fluorescence in situ hybridisation (FISH) were nick translated with biotin-14-dATP and hybridised in situ at a final concentration of 20 ng/μl to metaphases from the probands, their mothers, and normal controls. The FISH method was modified from that previously described by Callenet al,20 in that chromosomes were stained before analysis with both propidium iodine (as counterstain) and DAPI (for chromosome identification). Images of metaphase preparations were captured by a cooled CCD camera using the CytoVision Ultra image collection and enhancement system (Applied Imaging Int Ltd). FISH signals and the DAPI banding pattern were merged for figure preparation.
PHYSICAL MAPPING
YACs were grown in 100 ml of selective media (uracil, tryptophan) for 72 hours at 30°C. Liquid DNA as well as DNA in agarose blocks was prepared using standard protocols. YAC DNA in agarose blocks was digested (partially or completely) with four restriction endonucleases (BssHII, MluI,SalI, and SfiI) in buffers supplied by the manufacturer (New England Biolabs). Digested DNA was separated onto 1% agarose gel using a GeneLine (Beckman) instrument. Gels were blotted and probed with left and right vector arm probes, specific probes, and total human genomic DNA. The restriction map was constructed manually.
The cosmids were isolated by screening the LLNL X-chromosome specific cosmid library using the insert of the IMAGE clone corresponding to EST R97207. The clones were ordered from the UK HGMP Resource Center.
EST MAPPING
Query to the Unigene EST collection at the National Center for Biotechnology Information21 gave five different clusters of ESTs as potentially located in the critical interval. Corresponding IMAGE clones were ordered from the HGMP Resource Centre and their inserts hybridised to Southern blots containing theEcoRI digested Xq13 YACs. Two ESTs were mapped to the critical region. One of them (R97207, Hs.16177) was contained in YAC 4X62B8 which is the shorter YAC covering both inversion breakpoints. Refined mapping was subsequently achieved with this clone using pulsed field gel electrophoresis under the conditions indicated above.
Results
CASE REPORTS
Case 1
The proband, an 8 year old male, presented from birth to day 15 with hypotonia and poor feeding which was attributed to the mother taking phenobarbital during her pregnancy because of epilepsy. Global developmental delay was noted from 18 months. Sitting was acquired at 14 months and he could not walk or easily stand unaided at 18 months. When examined at 8 years, his size and OFC were normal. There was no facial dysmorphism but he had slight strabismus. Café au lait spots were present on the neck, the left buttock, and the right thigh in addition to a large, diffuse, hypopigmented region on the left thigh. The proband had a very poor level of comprehension and verbal abilities. His IQ was evaluated to be 51. CT scan gave normal results and fragile X syndrome was ruled out.
Case 2
The proband, a 10 year old male, was referred to a paediatrician at the age of 8 years because of severe learning difficulties and possible attention deficit disorder. He was the product of a normal pregnancy apart from maternal depression. He exhibited delayed milestones, walking at 16 months and using minimal language until the age of 3. The mother had no learning difficulties at school. There is a family history of several psychiatric illnesses among her relatives. The father had major difficulties at school and in particular seemed to have visual processing problems. He was coping well in a manual labouring occupation. During the consultation, it was notable that the proband had slow cognition and poor speech but he had no difficulties with concentration. He had mild dysmorphic features with a long, narrow palate, large ears, and scaphocephaly. His muscle tone was reduced. There was evidence of neuromaturational immaturity. Formal psychological assessment showed his verbal index to be 104, which is on the 61st centile, performance index was 69, which is on the 2nd centile, and full scale index was 85, which is on the 16th centile. The difference of 35 points between the verbal and performance index is significant (p<0.05) and is an uncommon disparity.
FISH AND PHYSICAL MAPPING IN Xq13
Two inverted X chromosomes have been identified, one with a pericentric inversion (INV1) 46,Y,inv(X)(p11.1q13.1) and the other with a paracentric inversion (INV2) 46,Y,inv(X) (q13.1q28). Both male carriers of the inverted chromosomes suffer from a mild form of non-specific mental retardation (see above). Identical rearranged chromosomes were found in their mothers, who are intellectually normal. Initial cytogenetic analysis indicated that both inverted chromosomes break in Xq13 (one of each inversion breakpoint). The studied X chromosome region and both chromosomal rearrangements are schematically represented in fig 1A.

(A) Schematic diagram of the X chromosome with a physical map of the Xq13.1 breakpoint region. The position of the markers from DXS453 to PHKA1 is not drawn to scale. DXS6673E is a gene isolated from a female patient with a translocation X;13.13 IL2RG, GJB1, p54nrb, CCG1, RPS4X, and PHKA1 are genes that have been isolated and characterised.19 ,28 ,30-32 YAC clones are represented by solid lines below the markers. YACs 8003 and A76C5 are from Lafrenière et al.32 The other YAC clones were isolated from the ICI, CEPH, or ICRF YAC libraries (Villard et al, unpublished data). (B) Long range restriction map of the region using four restriction endonucleases, BssHII, MluI, SalI, and SfiI. YAC and cosmid clones isolated and mapped to the region are shown underneath. The two inversion breakpoints (INV1 and INV2) map to two different intervals separated by a maximum of 250 kb. The localisation of a group of ESTs (the reference EST being R97207, Hs.16177) to the region just proximal to DXS131 is shown with a dotted line with an arrow at both ends. The size of the four SfiI restriction fragments detected in this region is indicated.
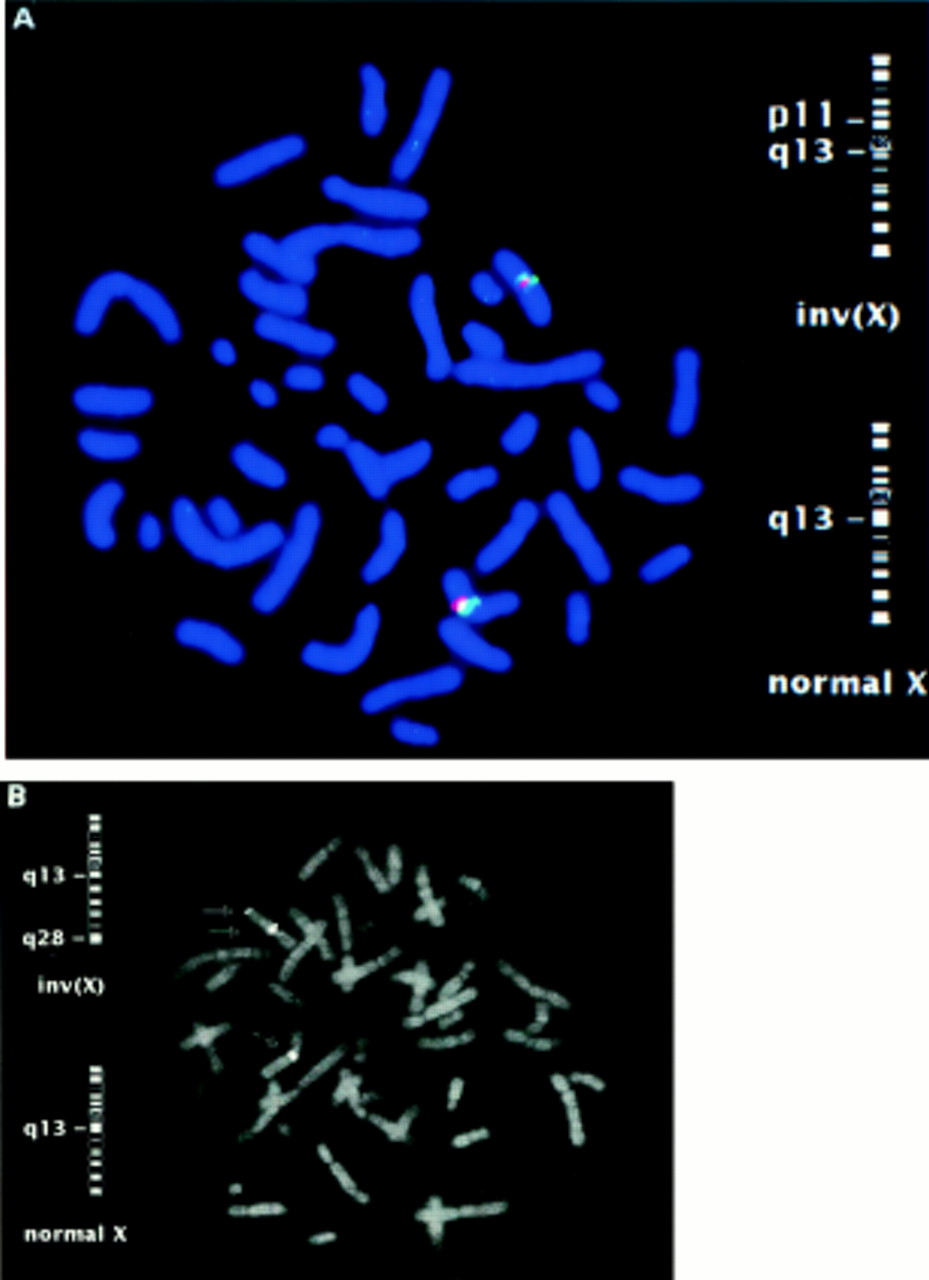
FISH mapping in the region of the common breakpoint in Xq13 showed that both chromosomes break in a region near DXS131 and DXS162 in Xq13.1. Several YACs from this region were found to cross one or both Xq13.1 breakpoints suggesting two independent breakpoints close to each other. As shown in fig 1B, YACs 769_f_2, 917_e_6 and 4X62B8 cross the INV1 breakpoint. YACs 798_c_10 and 4X62B8 cross the INV2 breakpoint. FISH results using YAC 4X62B8 which crosses both the INV1 and the INV2 breakpoints are shown in fig 2.

Fluorescence in situ hybridisation results obtained in the patients’ mothers’ metaphases showing the YAC clone 4X62B8 crossing the INV1 (A) and INV2 (B) breakpoints together with the signal obtained on the normal X chromosome. In the case of the INV1 breakpoint, the X chromosome alpha satellite probe and the YAC probe were cohybridised.
All YACs were subsequently used to build a long range restriction map of the breakpoint region (fig 1B and data not shown). To refine the breakpoint region further, the EST R97207 was used as a probe to isolate smaller clones by screening an X chromosome specific cosmid library. Positive cosmid clones 7G15, 10L5, and 52B14, which mapped to the region (see below), crossed the INV1 breakpoint. Cosmid clones 3J12, 4N22, and 4P14 are positive for DXS162 and were found to lie in between the two breakpoints. Taken together, these data show that both chromosomal breakpoints are located within a 250 kb genomic fragment in Xq13.1.
EST MAPPING IN Xq13
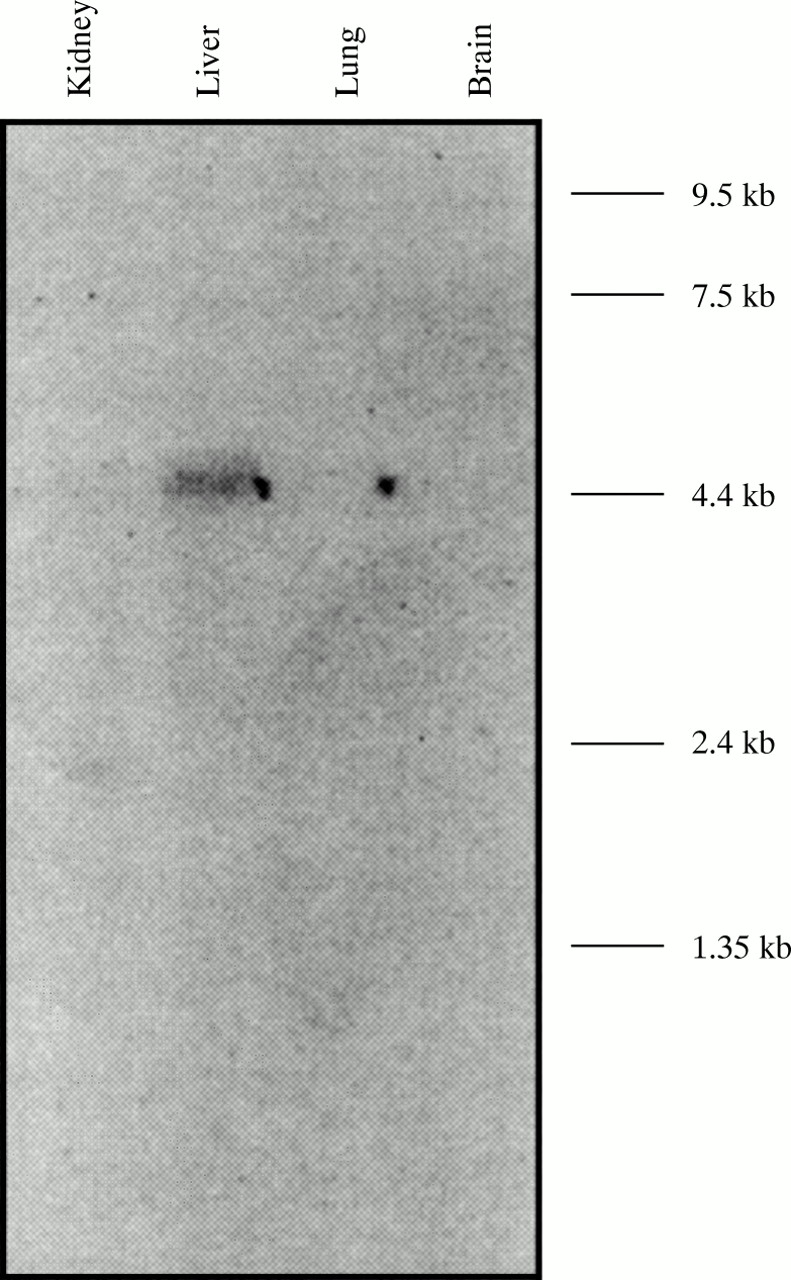
In the process of constructing a detailed transcriptional map of the proximal long arm region of the human X chromosome (Villardet al, unpublished data), a group of nine ESTs represented by the EST R97207 (Unigene cluster Hs.16177) was mapped to the Xq31.1 region and more accurately to theBssHII-MluI restriction fragment immediately proximal to DXS131 (fig 1B). All the ESTs belonging to this group of putative transcribed sequences originate from a fetal liver/spleen cDNA library. The R97207 clone was used to probe a human fetal northern blot where it detected a 4.5 kb fetal liver specific transcript (fig 3). The isolation of the full length corresponding cDNA is currently under way.
{kind=link}
{kind=link}
{kind=link}
Human fetal multiple tissue northern blot hybridised with the purifed insert of the EST clone R97207 showing the 4.5 kb transcript that is detected only in liver.
Xp11 AND Xq28 BREAKPOINTS
Regarding the opposite inversion breakpoint of INV1 and INV2, these were mapped to the Xp11 and Xq28 regions, respectively. Several YACs and cosmids were used for FISH mapping. The INV1 breakpoint was localised to the DXS14-DXS390 region in Xp11 and the INV2 breakpoint to the GABRA3-BGN region in Xq28 (results not shown).
Discussion
The study of balanced chromosomal rearrangements associated with an abnormal phenotype has allowed the isolation of numerous disease causing genes.22 This approach is particularly suitable for the study of non-specific X linked mental retardation since genetic linkage analysis is of limited use, mainly owing to the genetic heterogeneity underlying MRX and the broad regional localisations obtained.
We report here two unrelated male patients with inversions of the X chromosome. One is a pericentric inversion inv(X)(q13.1q28) and the other a paracentric inversion inv(X) (p11.1q13.1). Both boys are affected with non-specific X linked mental retardation and both inverted chromosomes have a common area of breakage in Xq13.1. We initially focused our attention on this region of the human X chromosome to determine if a single gene defect could be the cause of both phenotypes. FISH mapping and pulsed field gel electrophoresis restriction mapping on YAC clones was carried out to determine precisely the position of each of the breakpoints in Xq13.1. We have shown that both breakpoints are located in a 250 kb genomic fragment near DXS131-DXS162. This small chromosomal region could potentially contain a large gene interrupted by both breakpoints. To date, no known genes have been localised to the area of common breakage. The only gene reported to map to the DXS131-DXS162 interval,ne-dlg3, appears in fact to map more centromeric (close to DXS453) (Villard et al, unpublished data) in contrast to its published localisation.23 We took advantage of the ongoing EST mapping initiative21 and have shown that one of the ESTs broadly mapped to this region of the human X chromosome (EST R97207) was mapping within the critical interval, close to DXS162. Northern analysis showed that the corresponding transcript is liver specific. Although liver specific genes have been shown to be involved in some mental retardation phenotypes, the expression data obtained using this EST make it a poor candidate to be involved in the MRX phenotype of the studied inversion patients. We are isolating the whole 4.5 kb transcript to determine its position with respect to both breakpoints, because of a potential secondary effect of abnormal accumulation of metabolites in brain which may cause an MR phenotype (for instance, phenylalanine hydroxylase and phenylketonuria24). Additional transcribed sequences will also be isolated from the interval using different approaches (exon trapping, cDNA selection, and ultimately genomic sequencing).
The isolation of a gene interrupted by both breakpoints will not only uncover the molecular basis of the phenotype(s) of the inversion patient(s) but will also provide a candidate gene to test in the numerous other MRX families whose linkage intervals overlap this region of the X chromosome. The finding that oligophrenin-1 was mutated only in a single MRX family6 (MRX60) raises the possibility that a major MRX gene may exist in this region of the chromosome that still has to be isolated.
Although the Xq13.1 region is the most likely location for an MRX gene interrupted by inversion breakpoints in both patients, the possibility that each of the phenotypes observed in the patients is a consequence of a gene defect occurring at the other side of the corresponding inverted region cannot be excluded. The two opposite inversion breakpoints were mapped to small intervals between loci DXS14 and DXS390 (Xp11.1, case 1) and GABRA3-BGN (Xq28, case 2), respectively. Interestingly, the four inversion breakpoints described here (Xp11.1, Xq13.1, and Xq28) differ from those described by Sloan-Benaet al 15 (Xp11.2 and Xq21.3), thus excluding the possibility of a common gene interrupted by these breakpoints. Although the hypothesis of a “disease gene at the breakpoint” would be the simplest one, more and more evidence is being accumulated supporting the existence of different mechanisms underlying disease with chromosomal rearrangements outside genes and their regulatory regions known as position effect (reviewed by Lupski25 and Kleinjan and van Heyningen26). Potential contribution of other, autosomal gene(s) to the phenotype of the two inv(X) patients cannot be formally excluded either, at least until a gene interrupted by one of the breakpoints is identified.
An ever increasing number of transcription units are being placed on the integrated maps of human chromosomes27 (currently over 30 000, http://www.ncbi.nlm.nih.gov/genemap/). This resource together with the growing knowledge of the molecular pathways involved in mental retardation10 and the identification of MRX associated with X chromosome rearrangements could significantly speed up the isolation of the remaining MRX genes.
Acknowledgments
We would like to thank the families and the patients for their cooperation, John Mulley for critical reading of the manuscript, David Callen for help with case 2, Daniela Toniolo for the Xq28 probes, and Sarah McDonnell and Shaun Barnett for technical assistance. The authors are grateful to the UK HGMP Resource Centre for providing most of the YACs and IMAGE clones used in this study, and the Sanger Center for providing the PAC clones. This work was supported by the INSERM program PARMIFR, the PROGRES network, by the National Health and Medical Research Council of Australia, the Adelaide Women’s and Children’s Hospital Research Foundation, and the Association Française contre les Myopathies (AFM).
References
Footnotes
-
↵* The first two authors contributed equally to this work