Article Text

Abstract
Background BRCA1 and BRCA2 are the two principal tumour suppressor genes associated with inherited high risk of breast and ovarian cancer. Genetic testing of BRCA1/2 will often reveal one or more sequence variants of uncertain clinical significance, some of which may affect normal splicing patterns and thereby disrupt gene function. mRNA analyses are therefore among the tests used to interpret the clinical significance of some genetic variants. However, these could be confounded by the appearance of naturally occurring alternative transcripts unrelated to germline sequence variation or defects in gene function. To understand which novel splicing events are associated with splicing mutations and which are part of the normal BRCA2 splicing repertoire, a study was undertaken by members of the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium to characterise the spectrum of naturally occurring BRCA2 mRNA alternate-splicing events.
Methods mRNA was prepared from several blood and breast tissue-derived cells and cell lines by contributing ENIGMA laboratories. cDNA representing BRCA2 alternate splice sites was amplified and visualised using capillary or agarose gel electrophoresis, followed by sequencing.
Results We demonstrate the existence of 24 different BRCA2 mRNA alternate-splicing events in lymphoblastoid cell lines and both breast cancer and non-cancerous breast cell lines.
Conclusions These naturally occurring alternate-splicing events contribute to the array of cDNA fragments that may be seen in assays for mutation-associated splicing defects. Caution must be observed in assigning alternate-splicing events to potential splicing mutations.
- BRCA2
- breast cancer
- tumor suppressor
- alternate splicing
Statistics from Altmetric.com
Introduction
Breast cancer is among the leading causes of cancer deaths in women worldwide (http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx). While the majority of breast cancers are sporadic, some are associated with inherited ‘mutations’ (pathogenic variants) in the BRCA1 (MIM 113705) or BRCA2 (MIM600185) tumour suppressor genes. Indeed, BRCA1/2 pathogenic variants are the strongest genetic predictors of familial breast/ovarian cancer syndrome. Accordingly, individuals with strong family histories of breast and/or ovarian cancer are advised to seek genetic testing for BRCA1/2 sequence variants, results of which can be used to inform disease screening, disease prevention and disease treatment options.
BRCA1/2 clinical testing typically involves sequence analysis of all coding exons and adjacent intronic sequences. Though there are several known pathogenic missense alterations for each gene, most pathogenic variants are ‘protein truncating’ and include frame-shifting insertion/deletions, nonsense substitutions and base substitutions that alter normal physiological splicing. An ongoing challenge in clinical genetic testing is characterisation of DNA sequence variants of uncertain clinical significance (VUS).1 ,2 Effective genetic counselling requires that the pathogenicity of these variants be determined.3
To address this problem, the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) is exploring a series of approaches to characterise the potential pathogenicity of BRCA1/2 VUS identified during clinical screening, including those that may affect splicing.2 ,4 ,5 These investigations can be complicated by the observation that both BRCA1 and BRCA2 genes are transcribed into multiple naturally occurring mRNA splice isoforms independent of any genetic variants, and that these isoforms can vary in expression level between different individuals. Thus, presumed aberrant transcript patterns seen in the presence of a VUS, in some cases, could be unrelated to the VUS and thus not necessarily pathogenic.
In order to accurately interpret clinical splicing assays, it is necessary to provide an overview of the naturally occurring alternate-splicing events. ENIGMA collaborators have recently demonstrated the value of comprehensive approaches to catalogue previously unrecognised naturally occurring BRCA1 mRNA transcripts.6 Here, we report a complementary study conducted to provide the first systematic catalogue of human alternate BRCA2 mRNA splicing events. We have performed experiments in different blood-related RNA sources (from both healthy controls and patients with breast cancer with or without known BRCA1/2 pathogenic variants), breast cancer cell lines and non-breast cancer cell lines to show that the isoforms overlap in their expression profile across clinically relevant sources of mRNA.
Materials and methods
Phase Ia: BRCA2 alternate-splicing events reported by participating ENIGMA laboratories
Seven contributing ENIGMA laboratories used various cDNA amplification strategies to identify naturally occurring alternate-splicing events in controls who do not have breast cancer, from whole blood leucocytes (LEUs), ficoll-isolated peripheral blood mononuclear cells (PBMCs), primary cultures of stimulated peripheral blood lymphocytes (PBLs) and lymphoblastoid cell lines (LCLs), and one healthy breast tissue (BREAST) (see figure 1, table 1 and online supplementary table S1). Phase Ia was a collection of independent observations by individual ENIGMA participating groups, each using samples and methodologies available to those particular laboratories prior to the coordinated effort. The contributions of each group are described in online supplementary table S1. Briefly, cDNAs were generated with BRCA2 cDNA-specific primers or random hexamers/oligo dT, and these were amplified by PCR with BRCA2-specific primers designed at each contributing laboratory (all sequences are available upon request). Products were visualised on agarose gels or with capillary electrophoresis. In most cases, splicing isoforms were verified by sequence analysis at the originating institution. Samples and methods have been described in detail elsewhere.6 Details are presented in the ‘Results’. In brief, 22 BRCA2 alternative splicing events were identified. Phase Ia was conducted to detect BRCA2 splicing events, not to address quantitative aspects. Yet visual inspection of capillary electrophoresis experiments allowed us to annotate four splicing events (Δ3, Δ6q,7, Δ12 and Δ17,18) as predominant, a qualitative annotation indicating that they represent a non-negligible/substantial fraction of the total alternate-splicing events, particularly if compared with other (minor) events (see representative examples in figure 2). Predominant events are associated with much higher detection rates than minor events, regardless of the samples analysed (table 1).
BRCA2 alternate mRNA splicing events identified in phase Ia

Schematic diagram showing the process of identification and validation of alternate-splicing events of BRCA2 mRNA. Alternate-splicing events submitted by contributing Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) members were identified in healthy controls from whole blood leucocytes, peripheral blood mononuclear cells, (primary cultures of stimulated blood lymphocytes (and lymphoblastoid cell lines (LCLs) (phase Ia). cDNA exon scanning was performed using mRNA prepared from MCF7, MCF10A and 184A1 (phase Ib). The two alternate-splicing events added between phases I and II are predicted events ▾5p (from a previous study) and Δ9–11, reported in ENCODE (see text).

Representative examples ofagarose and capillary electrophoresis analyses performed in phase Ia. BRCA2 alternative spliced events were detected by RT-PCR with forward and reverse primers located in the indicated exons. Size differences between full-length and alternative splicing transcripts are indicated in nucleotides (nt). We indicate both the difference as determined by capillary electrophoresis analyses (size-calling performed with GeneScan software) and (in brackets) the expected difference based on BRCA2 exons size. Note the qualitative differences (in terms of relative signal compared with the full-length) between predominant (Δ3, Δ6q,7, Δ12, Δ17,18) and minor (Δ2 Δ5, Δ5_7, and Δ12,13) events. Predominant events were consistently detected in ethidium bromide-stained agarose gels. Note that BRCA2 Δ12 is analysed with different RT-PCR reverse primers in capillary (left) and agarose (right) electrophoresis.
Supplementary table
Twenty-two candidate isoforms reported by ENIGMA collaborators were compiled and submitted to the University of Chicago where their expression in LCLs and breast normal and tumour cells was tested by isoform-specific RT-PCR and sequencing analyses as described below. Two additional potential alternate-splicing events not seen in phase Ia were also tested: Δ9–11, which was reported in the ENCODE RNA-Seq data set from the California Institute of Technology http://www.ncbi.nlm.nih.gov/geo/info/ENCODE.html, and ▾5p, which was predicted from a spliceogenic variant reported in a previous study.5
Phase Ib: alternate splice product detection and sequencing
An exon-scanning strategy was developed at the University of Chicago to identify potential abundant BRCA2 splicing isoforms. This strategy was designed independently of the design or results in phase Ia. Briefly, 18 primer pairs were designed to amplify portions of the BRCA2 cDNA flanking exons 2 (alone), 3 (alone), 4–6, 5 and 6, 5–7, 9 alone, 12 and 13, 13 alone, 14 and 15, 15 alone, 16 alone, 16 and 17, 18–20, 19 and 20, 19–22, 23 and 24, 23–25 and 23–26. These primers are able to generate both full-length RT-PCR products and any product resulting from intervening exon-skipping or alternate splice-site choice. RT-PCR products were run on 1% agarose gels and visualised with ethidium bromide staining and UV transillumination according to standard protocols (figure 3A). Gel-isolated cDNA fragments were sequenced using the forward or reverse RT-PCR primer using standard protocols. The exon-scanning strategy was performed on mRNA prepared from MCF7, MCF10A and 184A1 cell lines. This strategy covered all cDNA fragments except those containing exons 10 (1116 bp) or 11 (4932 bp) because their large sizes did not permit convenient analysis of full-length large-exon cDNA fragments.

BRCA2 alternate splice products detected by scanning RT-PCR (phase Ib). (A) Primers flanking one or more exons were designed to detect alternate splice products. The coloured horizontal bars represent any region of a BRCA2 mRNA molecule with any three exons (arbitrarily coloured red, blue and green). Coloured arrows represent RT-PCR and cDNA PCR primers. The top figure represents a full-length mRNA molecule according to the reference sequence (National Center for Biotechnology Information accession number NM_000059), and the bottom figure represents a fragment of a corresponding BRCA2 mRNA that has undergone an alternate-splicing event (in this case, exon skipping). Examples of alternate-splicing event detection are shown for Δ3 (B) and Δ12 (C). Full-length RT-PCR products (upper bands) and exon-deleted isoforms (lower bands) are shown next to electropherograms demonstrating the corresponding exon–exon junctions. The highest molecular weight band in (B) and (C) likely represents a heteroduplex containing some strands of Δ3 and Δ12 and full-length RT-PCR products, respectively.
Phase II: isoform-specific RT-PCR primer design
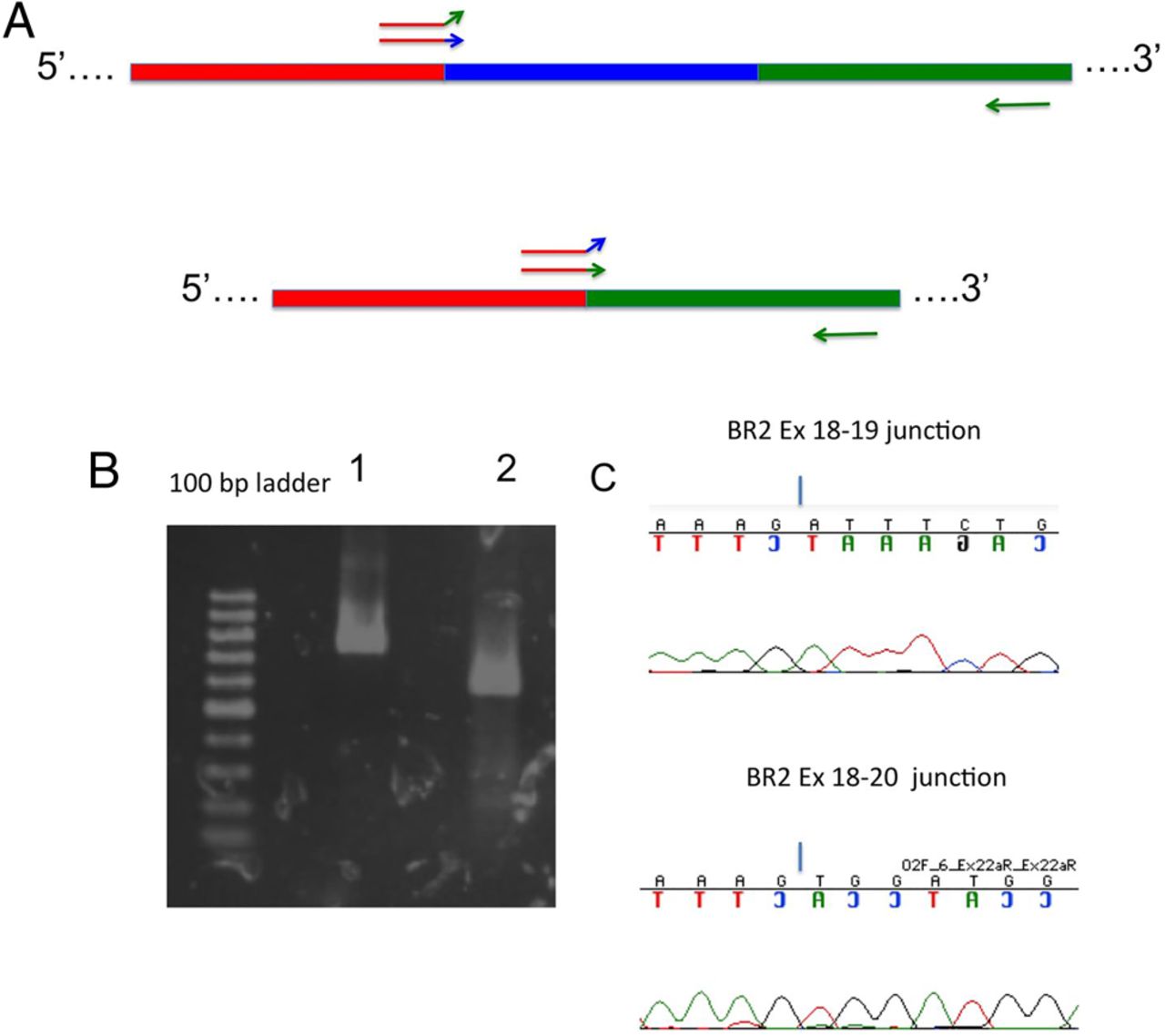
To validate potentially low-abundance BRCA2 alternate-splicing events with utmost specificity, we designed exon boundary junction-spanning isoform-specific RT-PCR primers. We used these to verify specific candidate splicing isoforms detected in phase I without competition from full-length splicing products.7–9 These isoform-specific primers contained 3–5 bases of 3′ sequence designed to hybridise with the 5′ end of the adjacent exon in either the full-length BRCA2 mRNA or in an isoform resulting from exon skipping (see figure 4 and supplementary table S2). As we use a one-step RT-PCR method (see below), the reverse PCR primer is also the primer used by the RT to generate the single-stranded cDNA. In our experiments, when the reverse primer is the isoform-specific primer, it often amplifies both full-length and alternate-splicing events, possibly because nuclease activity trims back the isoform-specific 3′ end of the primer or because of some kind of non-specific binding event. However, this occurs less frequently when the isoform-specific primer is the forward RT-PCR primer. We have therefore designed all isoform-specific RT-PCR primers as forward primers (see online supplementary table S2).

BRCA2 alternate splice products are amplified by isoform-specific RT-PCR. (A) Illustration of isoform-specific RT-PCR primer design, with an upstream primer specific for a portion of a full-length cDNA including red, blue and green exons (top) or specific for an exon-skipping cDNA that lacks the blue exon (bottom). Note the forward primer with the green sequence 3′ end is unable to amplify the full-length cDNA fragment and the forward primer with the blue 3′ sequence end is unable to amplify the exon-skipping product. An example of isoform-specific RT-PCR is shown. (B) An agarose gel shows RT-PCR products generated using primers for the exon 18–19 junction (lane 1) or the exon 18–20 junction unique to the Δ19 isoform (lane 2). (C) Corresponding electropherograms confirming the identities of the RT-PCR products.
Patient samples and cell lines
A description of the patient samples and cell lines used in phase II is provided in the online supplementary materials and methods.
Supplemental material
RT-PCR
mRNA was prepared from cells using RNeasy Mini Kit (Qiagen) according to the manufacturer's protocol. In contrast to other studies of alternate-splicing events, no inhibitors of nonsense-mediated mRNA decay (NMD) were used in phase II. RT-PCR was performed using the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Life Technologies) according to the manufacturer's protocol. PCR conditions were empirically optimised for each primer pair using a standard set of mRNAs (10–50 ng per reaction) prepared from select LCLs and breast cell lines.
Results
BRCA2 alternate-splicing isoforms reported by participating ENIGMA laboratories (phase Ia)
Seven groups contributed reports totalling 22 unique alternate BRCA2 splicing events detected by RT-PCR in mRNA samples prepared from various cell types (figure 1 and table 1). All splicing events were supported by direct sequencing evidence, with the only exceptions of Δ5,6; Δ6; Δ12,13; and Δ23p, which were predicted from capillary electrophoresis (EP) size-calling. All events were detected by capillary EP, but only eight events (36%) were detectable by conventional agarose gel electrophoresis/ethidium bromide (EtBr) staining. We were able to detect up to 15 events (68%) in LEU samples, 17 (77%) in PBMCs samples, 18 (82%) in PBL samples and 22 (100%) in LCL samples. We tested 21 splicing variants in RNA extracted from one healthy breast tissue (BREAST), detecting 10 (48%). The low value is likely related to the fact that each alternate-splicing event was tested only once due to the scarcity of biological material (see table 1 for further details).
Exon scanning (phase Ib)
Only three isoforms (Δ3, Δ6q7 and Δ12) were detected unambiguously by exon-scanning RT-PCR, followed by conventional EtBr-stained gel electrophoresis (a low-sensitivity detection method). The data nonetheless confirm Δ3, Δ6q7 and Δ12 as major alternative splicing events.
Specificity of isoform-specific RT-PCR primers
To validate and survey all 22 alternate-splicing events reported in phase I, an isoform-specific RT-PCR strategy was devised to amplify corresponding alternate cDNAs (see below). Specificity for individual alternate-splicing event products is determined by several bases at the 3′ ends of forward PCR primers that span splicing junctions being investigated. One potential source of non-specific cDNA amplification by this method is the possibility of a ‘loop-out’ structure formation by the exon junction-spanning isoform-specific primer that could result in an exon-skipping artefact (see online supplementary figure S1). This is unlikely to be a major contributor to the detection of isoforms using isoform-specific RT-PCR used in this study because of two sets of observations. First, most alternate-splicing events were not detected in every experiment, thus providing negative internal controls for most potential loop-out products. Specifically, of the 24 isoform-specific primers designed to identify exon-skipping isoforms, only one (BRCA2 ex 2–4 F, designed to identify the BRCA2 Δ3 isoform) amplified a product in every cell type examined. In our experiments, BRCA2 Δ3 is easily detected with conventional flanking RT-PCR primers not subject to loop-out artefact conditions. All other isoform-specific primers designed to identify exon-skipping isoforms failed to detect the target isoform in at least one cell line, whereas all corresponding control primers (ie, those designed amplify to full-length BRCA2 cDNA) amplified the correct RT-PCR product in all cell lines. Thus, for these primers, starting conditions for RT-PCR were not sufficient to generate exon-skipping isoforms by looping out or other false priming events. Indeed, a loop-out structure is predicted to be energetically unfavourable given the very short exon-spanning 3′ sequence of the isoform-specific primer. Second, we tested the ability of several isoform-specific RT-PCR primers to generate forced loop-out RT-PCR products. This was done by using exon-skipping isoform-specific RT-PCR primers in PCRs with gel-isolated full-length cDNA products as templates. Under these conditions, exon-skipping cDNAs could only be generated by spanning and amplifying a loop-out intermediate. We tested 12 isoform-specific forward RT-PCR primers representing exon-skipping isoforms for their ability to generate a forced loop-out product against a full-length template (see online supplementary figure S1). Also, 6 of the 12 were unable to force the generation of exon-skipping products from the full-length template. The other six were able to generate exon-skipping products when provided with large amounts of starting template. The primer that could generate a forced loop-out amplification with the smallest amount of input template was BRCA2 ex 11–14 F (designed to amplify the BRCA2 Δ12,13 splicing isoform). This primer was able to amplify a BRCA2 Δ12,13 product from 0.01 ng of full-length cDNA template, but not from 5×10–5 ng of cDNA, an amount over a hundred fold in excess of the corresponding full-length cDNA in the first round of an RT-PCR reaction using our conditions, assuming 100 ng RNA contains roughly 7×104 copies of a typical rare message.10 We therefore conclude that exon-skipping cDNA isoforms are unlikely to be artificially generated by isoform-specific primers under our RT-PCR conditions.
Supplementary figure
Isoform-specific RT-PCR amplification and sequence analysis (phase II)
The naturally occurring BRCA2 splicing isoforms reported by contributing institutions were collected as described in ‘Materials and methods’ for phase Ia (table 1). These were then validated at the University of Chicago using isoform-specific RT-PCR in MCF7, HCC1937, BT20, MCF10A, 184A1, 184B5 and at least 10 LCLs. Breast cell lines were included to address whether major differences in splicing events exist between clinically accessible blood-derived cells and cells derived from tissue affected by BRCA2-associated disease; all 24 alternate-splicing events assayed were detected in the examined cell lines with variable frequencies, ranging from 29% (Δ9–11) to 100% (Δ3) positive samples (table 2) and validated by sequence analysis. Representative cDNA fragments identified by scanning RT-PCR are shown in figure 3B, and an example of an isoform confirmed by isoform-specific RT-PCR is shown in figure 4B. Only 7/24 (29%) of the naturally occurring splicing events (namely, BRCA2 exon-skipping alternate-splicing events Δ3; Δ5; Δ6; Δ5,6; Δ12; Δ17,18 and Δ18) were reported previously in the literature (table 2). These were described as low-level background exon-skipping events in negative control samples used to characterise exon-skipping events associated with pathogenic sequence variants.11–17
BRCA2 alternate mRNA splicing events detected during phase II
Of the 24 isoforms confirmed by isoform-specific RT-PCR, there are 5 (▾5p, Δ6q7, ▾20A, Δ22–23p and Δ23p) that are not simple exon-skipping isoforms (figure 5). ▾5p represents an alternate splice acceptor site and includes 23 bases of intron 4 appended to the 5′ end of exon 5. Δ6q7 lacks exon 6 almost completely, except for two nucleotides (TG), and lacks exon 7 completely. ▾20A represents a cryptic exon, in which a 64 base sequence from intron 20 is inserted between exons 20 and 21. Δ23p and Δ22,23p also represent an alternate splice-site acceptor. In this case, however, the acceptor lies within exon 23, truncating it by 51 bases on the 5′ end.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structures of five BRCA2 alternate splice products including cryptic splice donor/acceptor sites. (A) Δ6q7, which contains an exon 6-derived TG dinucleotide between flanking exons 5 and 8. (B) ▾5p, which results from a cryptic intronic splice acceptor site 23 bases from the 5′ end of exon 5. (C) ▾20A results from a cryptic exon within IVS 20. (D) Δ23p and Δ22,23p both use a cryptic splice acceptor site within exon 23.
The ▾5p finding was assayed on the presumption that it was a possible splicing event. In an unrelated study,5 an isoform containing 18 intronic nucleotides upstream exon 5 was reported in a subject carrying a c.426–12_c.426-8delGTTTT that results in the use of a cryptic acceptor site. The corresponding isoform, using the same acceptor site in a wild-type allele, would be the ▾5p alternate-splicing event described here. Although the ▾5p splicing event was not observed previously in RNA samples from LCLs of BRCA1/2 pathogenic variant carriers or controls5 or by contributing members for phase Ia of this study, the isoform-specific RT-PCR analysis revealed this splicing event in 5/10 LCLs, BT20, MCF10A and 184A1 (table 2).
Another splicing event, Δ9–11, was not tested by contributing laboratories during phase I but was assayed because it appeared in the ENCODE RNASeq data. This splicing event was identified by isoform-specific RT-PCR in 2/11 LCLs as well as BT20 and 184A1.
Several BRCA2 cDNA fragments representing alternate-splicing events were detectable in most cell lines examined, though not all appeared in every cell type tested. For example, Δ3–7, Δ6q7 and ▾20A were all detectable as phase II agarose gel bands from isoform-specific RT-PCR in all the breast cell lines tested and the majority, but not all, of LCLs. In contrast, Δ9–11 was seen in only 2/6 breast cell lines and only 2/11 LCLs, while Δ20 was observed in only one breast cell line (184A1), even though it was detected in the majority of LCLs tested (table 2). Similarly, some cell lines produced detectable levels of most BRCA2 alternate-splicing event cDNA fragments, while others did not. For example, in this survey, all alternate-splicing events except Δ4 were detected in 184A1, while only 11/24 mRNA splicing isoforms were detected in 184B5, a cell line derived from the same individual as 184A1. These cell types are karyotypically and genetically similar, but not identical. Both are p16 negative, wild type for RB1 and originally wild type for TP53 though some 184A1 lineages have developed deficiencies in p53 functions.18 It is not clear whether different synthesis and degradation rates of isoforms resulting from alternate-splicing events are inherent to these cell lines or are influenced by subtle differences in cell growth and RNA preparation and storage conditions. However, it is important to note that the expression of many alternate-splicing events can be highly variable within and between cell types, which is an important consideration when interpreting splicing assay results in connection with potential molecular effects of a VUS. Collectively, these observations are consistent with the view that many alternate-splicing isoforms may be sensitive to cell growth conditions and RNA preparation and storage conditions such that expression patterns may show variable levels between experiments. It is also possible that some cDNAs are subject to stochastic PCR amplification, as we have demonstrated previously for BRCA1 splicing events.6
Discussion
Clinical sequencing of BRCA1 and BRCA2 is recommended to anyone with a strong family or personal history of breast/ovarian cancer. To provide the most complete information for genetic counselling, it is essential to describe the likely clinical significance of VUS identified during screening. RT-PCR analysis of any variant that could affect splicing is a powerful and efficient tool for identifying high-frequency exon skipping or intron retention events in mRNA that is easily prepared from peripheral blood or derivatives thereof. Cases where mutant alleles cause complete exon skipping typically correspond to pathogenicity. However, it is not yet possible to know how much reduction of normal message accumulation constitutes pathogenicity, nor do we know whether any alternate splice variants might result in dominant negative effects on normal gene activity. Addressing these questions will require further research.
The purpose of this study is to address a previously underappreciated challenge to interpreting splicing assays: the frequent occurrence of naturally occurring mRNA transcripts that can be mistaken for variant-associated splicing defects. In some cases, a potential spliceogenic variant might have modest effects on splicing and merely increases the frequency of a naturally occurring alternate mRNA splicing event, maintaining the expression of full-length mRNA. In such cases, the remaining full-length mRNA could provide some BRCA2 function. This result is especially important as numerous BRCA2 splicing ‘mutations’ have been reported, supported at least in part by the appearance of ‘novel’ RT-PCR products reported here as potential background isoforms. As shown in table 3, 17/24 naturally occurring splicing isoforms reported in this study have been previously reported to be aberrant transcripts associated with ‘mutations’ as determined by various assays (see references in table 3). In addition, the analysis must capture properly the alternative splicing existing in the region of interest to avoid misinterpretations, as illustrated previously for BRCA1 alterations in exon 9 and BRCA1 splicing isoform Δ9,10.19 For instance, it is not obvious whether the main outcome of impairing BRCA2 exon 17 splicing sites should be exon 17 skipping or exon17,18 skipping (since, like BRCA1 Δ9,10, BRCA2 Δ17,18 is already a predominant event in control samples). In this regard, the effect of the BRCA2 c.7806–9T>G (IVS16–9T>G) variant on splicing has been analysed by RT-PCR with a forward primer positioned in exon 15 and a reverse primer positioned in exon 18,20 therefore, excluding from analysis Δ17,18 transcripts. In our opinion, an analysis with a reverse primer located in exon 19 would be more informative.
Indeed, all BRCA2 exons except 4, 10, 11, 12 and 20 have been reported to be associated with at least one pathogenic variant near the intron/exon boundary and designated as pathogenic due to assumed/known splicing defects by the Breast Cancer Information Core (http://research.nhgri.nih.gov/bic). When applying splicing assays using RT-PCR to identify aberrant splicing products associated with DNA sequence variants near intron/exon boundaries, it is important to remember that such splicing events exist as part of the natural splicing process regardless of the presence of spliceogenic variants, and that varying amounts of full-length mRNA may be produced from the same allele.
In humans, the average number of protein coding transcripts per locus is roughly 4, according to the latest GENCODE release (V.24, http://www.gencodegenes.org/stats/current.html). Loci with >20 annotated splicing isoforms are very rare.21 Our data (up to 24 splicing events, including 4 predominant) suggest that BRCA2 is probably in the upper limit of an average locus for the number of alternate-splicing events. Yet, this is in contrast with BRCA1, a genomic locus similar to BRCA2 in size (≈81 kb vs ≈84 kb) and number of exons (23 vs 27), but with a notable higher level of alternative splicing (up to 63 splicing events, including 10 predominant), according to a recent study conducted by the ENIGMA consortium with a very similar methodology.6 This difference may reflect the well-established link between protein disorder (a structural feature of BRCA1 but not BRCA2 proteins) and high levels of alternative splicing.22 ,23
In summary, we have presented an overview of 24 alternate-splicing events associated with normal BRCA2 mRNA processing. These results are of importance to the design and interpretation of mRNA splicing assays, construct-based or of patient material, that are commonly used to assess whether VUS leads to aberrations that are phenotypically equivalent to a molecular null (ie, a gene deletion). Recent studies have shown that detection of alternate splice events can be highly variable between laboratories and is quite sensitive to variations in cell types, cell growth conditions, mRNA preparation and RT-PCR methodology.5 ,6 ,24 Recommendations have been made to help make methodologies and reporting of alternate-splicing isoforms more uniform, including inhibition of NMD, sequence confirmation, proper primer design, presentation of data from at least 10 control samples of the same cell type to reveal potentially rare alternate splice isoforms, quantifying the variant allele contribution to full-length mRNA isoforms and quantifying the level of ‘aberrant’ transcripts relative to the full-length transcript.24 These, however, are suggestions for achieving uniformity of alternate-splicing event detection and quantification, but not directly characterising the risk phenotype associated with a genetic variant in families. Moreover, disease-associated alternate-splicing events would likely be associated with loss of heterozygosity in the affected tissues. Thus, examination of relative levels of ‘normal’ and ‘aberrant’ transcript levels in blood derived may not reveal levels of exclusively ‘aberrant’ transcript levels in affected tissues. Where interpretation of a splicing aberration is not straightforward, the full phenotypic consequence of variants should preferably be assessed with additional evidence from multifactorial models that incorporate largely evidence based on the clinical characteristics of carriers of bona fide pathogenic variants.25 ,26
After the completion of this work, several contributing labs on this study reported identification of BRCA2 cDNA fragments with combined alternate-splicing events, ▾20A, Δ22 and ▾20A, Δ22,23. Others identified BRCA2 Δ11, BRCA2 Δ11–12 and Δ11–13 in wild-type controls while investigating the splicing effects of the IVS11+1G>C spliceogenic variant (table 2). We anticipate other alternate-splicing events and combinations will be identified over time and require characterisation.
Acknowledgments
The University of Chicago group is grateful to The Entertainment Industry Foundation, Ralph and Marion Falk Medical Research Trust and Breast Cancer Research Foundation. The authors thank the following personnel of INT: Fernando Ravagnani for providing biological samples, Claudia Foglia, Donata Penso and Maria Teresa Radice for technical assistance. They are grateful to the Spanish ISCIIII-Subdirección General de Evaluación y Fomento de la investigación (I+D+I 2008–2011) and Fondo Europeo de Desarrollo Regional-FEDER. The authors also thank Anna Tenés for technical support and the ICO Hereditary Cancer Program team led by Dr Gabriel Capella.
References
Footnotes
Contributors All participants in this study are university-associated members of the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium, and all have provided research samples, wet lab results, literature searches, data compilation and analysis, and critical manuscript reading. JDF designed the experiments executed by the team at the University of Chicago (funded by OIO), performed some of the experiments, all of the analysis and wrote the manuscript. Other experiments and critical manuscript reading were provided by the other members of the Chicago team: TY, BZ, GRdG (then visiting Chicago), SCA and KL. The members of Fondazione IRCCS Istituto Nazionale dei Tumori (the Milan team), headed by R, all participated in generating data and providing critical commentary, as did the members of the Fundación Pública Galega de Medicina Xenómica-SERGAS (Santiago de Compostela) team, AV, MS and AB. AB, working under the supervision of BW (University of Cologne and University Hospital Cologne), provided additional data. Both provided critical commentary. CH (Institut Curie and Université Paris Descartes) provided leadership and critical manuscript reading, as did LCW (University of Otago) and IL-P (Instituto de Investigación Sanitaria San Carlos (IdISSC)). MT (Odense University Hospital) provided special computational predictions as well as critical manuscript reading. The team from the QIMR Berghofer Medical Research Institute (the Brisbane team), directed by ABS (chair of the Splicing Working Group of ENIGMA), included MP, who performed extensive literature searches, and PW. All provided valuable data, critical manuscript reading and insight into the development of the project. MJB directed the efforts of the team from Maastricht University Medical Center (the Maastricht team) and was one of the intellectual founders of the project. The team included DT and DB who, along with MJB, provided raw data, data analysis and critical reading of this manuscript. Similar work was provided by DB (University of Southhampton). The Vall d'Hebron Institute of Oncology (VHIO) and Universitat Autonoma de Barcelona group (the Barcelona group) was directed by OD, and included GM and SG-E. All members provided samples, raw data and critical assessment of the manuscript. Likewise, CL (IDIBELL–Catalan Institute of Oncology) provided data and critical reading. Samples used in this study included those provided by kConFaB Investigators who are hereby acknowledged. The first phase of this project was directed by MdlH (Instituto de Investigación Sanitaria San Carlos (IdISSC)) who, along with GRdG, performed much of the data generation and all of the phase I data compilation. Both provided intellectual leadership, raw data and critical reading of the manuscript.
Funding The authors acknowledge the research grants and RTICC are initiatives of the Spanish Ministry of Economy and Innovation partially supported by European Regional Development FEDER Funds. This work was funded by the National Cancer Institute grant numbers CA-R01 89085-01A and SPORE P50 CA125183 (to OIO), US Army DOD Grant #BC990302 (to JDF), The Italian Association for Cancer Research (AIRC) grant number 11897 (to PR), the NHMRC Senior Research Fellowship number ID1061779 (to ABS), The Cancer Council Queensland grant number ID1086286 (to MP), the Spanish Instituto de Salud Carlos III research grant PI12/00539 (to MH), Red Temática de Investigación Cooperativa en Cáncer-RTICC research grants RD12/0036/006 and RD12/0036/008 (to MH an GRG), Miguel Servet contract numbers CP10/00617 (to SGE) and PI12/02585 (to OD), and the Asociación Española Contra el Cáncer, Spanish Health Research Fund, Carlos III Health Institute, Catalan Health Institute and Autonomous Government of Catalonia, Contract grant numbers: RD12/0036/008, PI10/01422, PI10/00748, PI13/00285, PIE13/00022, 2009SGR290 and 2009SGR283.
Competing interests None declared.
Ethics approval University of Chicago Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All raw data will be made available to any interested investigator