Article Text

Abstract
Background Fetal akinesia deformation sequence syndrome (FADS, OMIM 208150) is characterised by decreased fetal movement (fetal akinesia) as well as intrauterine growth restriction, arthrogryposis, and developmental anomalies (eg, cystic hygroma, pulmonary hypoplasia, cleft palate, and cryptorchidism). Mutations in components of the acetylcholine receptor (AChR) pathway have previously been associated with FADS.
Methods and results We report on a family with recurrent fetal loss, where the parents had five affected fetuses/children with FADS and one healthy child. The fetuses displayed no fetal movements from the gestational age of 17 weeks, extended knee joints, flexed hips and elbows, and clenched hands. Whole exome sequencing of one affected fetus and the parents was performed. A novel homozygous frameshift mutation was identified in muscle, skeletal receptor tyrosine kinase (MuSK), c.40dupA, which segregated with FADS in the family. Haplotype analysis revealed a conserved haplotype block suggesting a founder mutation. MuSK (muscle-specific tyrosine kinase receptor), a component of the AChR pathway, is a main regulator of neuromuscular junction formation and maintenance. Missense mutations in MuSK have previously been reported to cause congenital myasthenic syndrome (CMS) associated with AChR deficiency.
Conclusions To our knowledge, this is the first report showing that a mutation in MuSK is associated with FADS. The results support previous findings that CMS and/or FADS are caused by complete or severe functional disruption of components located in the AChR pathway. We propose that whereas milder mutations of MuSK will cause a CMS phenotype, a complete loss is lethal and will cause FADS.
- Clinical genetics
Statistics from Altmetric.com
Introduction
Fetal akinesia deformation sequence (FADS) (OMIM 208150), also known as Pena-Shokeir syndrome I or arthrogryposis multiplex congenita (AMC) with pulmonary hypoplasia, is a rare condition associated with malformations caused by reduced fetal movement (fetal akinesia). About 30% of affected individuals are stillborn and many live born infants survive only a short time due to complications of pulmonary hypoplasia.
FADS is characterised by arthrogryposis, fetal akinesia, intrauterine growth restriction, developmental abnormalities such as cystic hygroma, pulmonary hypoplasia, cleft palate, cryptorchidism, cardiac defects, intestinal malrotation and sometimes pterygia of the limbs. It shows phenotypic overlap with the lethal type of multiple pterygium syndrome (LMPS, OMIM 253290).1
FADS is most often inherited as an autosomal recessive trait,2 but X-linked or dominant inheritance have also been suggested.3 ,4 Several mutations have been described in FADS and/or LMPS, explaining around 24% of the cases, and involving genes in the motor neuron development and survival, genes encoding components of the neuromuscular junction (NMJ), adult skeletal muscle proteins, and fetal myostructural proteins.5 The mutations located in genes associated with the NMJ include the subunits of the acetylcholine receptor (AChR): CHRNA1 (OMIM 100690),6 CHRND (OMIM 100720)6 and CHRNG (OMIM 100730);7 and genes signalling with muscle, skeletal receptor tyrosine kinase (MuSK) in the prepatterning process: RAPSN (OMIM 601592)8 and DOK7 (OMIM 610285).9 Mutations in SYNE1 (OMIM 608441), also involved in the prepatterning process, leads to AMC,10 and mutations in CNTN1 (OMIM 600016), a neural adhesion and NMJ protein, have been described to cause a congenital lethal myopathy11 (figure 1).

Fetal akinesia deformation sequence (FADS) and/or multiple pterygium syndrome (MPS) are associated with genes involved in the neuromuscular junction (NMJ). Several genes (CHRNA1, CHRND, CHRNG, RAPSN, DOK7, CNTN1 and SYNE1) involved in the NMJ have previously been associated with FADS. One additional gene, muscle, skeletal receptor tyrosine kinase (MuSK) (OMIM 601296) was identified that can be used in prenatal testing or genetic analysis for NMJ disorders.
Other non-lethal diseases involved in the NMJ pathway include congenital myasthenic syndrome (CMS, OMIM 608931) and Escobar syndrome (OMIM 265000). CMS can be classified as presynaptic, synaptic or postsynaptic. Approximately 75% of CMS cases are postsynaptic, the majority of which are caused by AChR deficiency.12
The emergence of high throughput DNA sequencing technologies has made it possible to sequence more rapidly and less expensively the coding parts of a patient’s genome, to enable unbiased searching for novel disease genes and identify potentially damaging mutations. We performed whole exome sequencing (WES) of a family trio, where the parents had five affected fetuses/children with a severe lethal form of FADS and one healthy child. Diagnosis of the fetuses was unknown until identification of the disease-causing gene was completed. This study reports a novel lethal homozygous insertion in the gene encoding MuSK. The frameshift mutation leads to a premature stop codon and likely a non-functional protein, causing the FADS phenotype.
Methods
Patients
The parents were both in their 30s at the time of their first visit to the fetal medicine unit, Uppsala University Hospital, Sweden, in 2003. There was no known consanguinity, but a common ancestor cannot be excluded due to the parents deriving from the same geographical area. At the time, they had one previous pregnancy, which ended in early miscarriage. The pedigree of the affected family in 2014 comprised two miscarriages, five affected children/fetuses, and one healthy child (figure 2A). The pregnancy in 2003 had a normal second trimester scan at their home clinic. In gestational week 23, the woman presented with high symphysis fundal measures of uterus at a standard check up with her midwife. Ultrasound examination revealed polyhydramnion and fetal hydrops and no fetal movements (figure 2B). The following examinations were conducted at the fetal medicine unit, Uppsala University Hospital. Table 1 shows the outcome of the pregnancy in 2003 and four additional pregnancies (2005, 2005, 2007, 2010), with fetuses/children presenting with findings consistent with FADS (figure 2C). All fetuses/children showed akinesia and joint contractions at ultrasound examination, and autopsy confirmed muscle atrophy and multiple joint contractions. In 2008 a healthy boy was born. Genomic DNA was extracted from EDTA blood (parents), saliva (healthy sibling), and lung tissue (affected fetuses) using standard methods.
Outcome of the eight pregnancies in the family
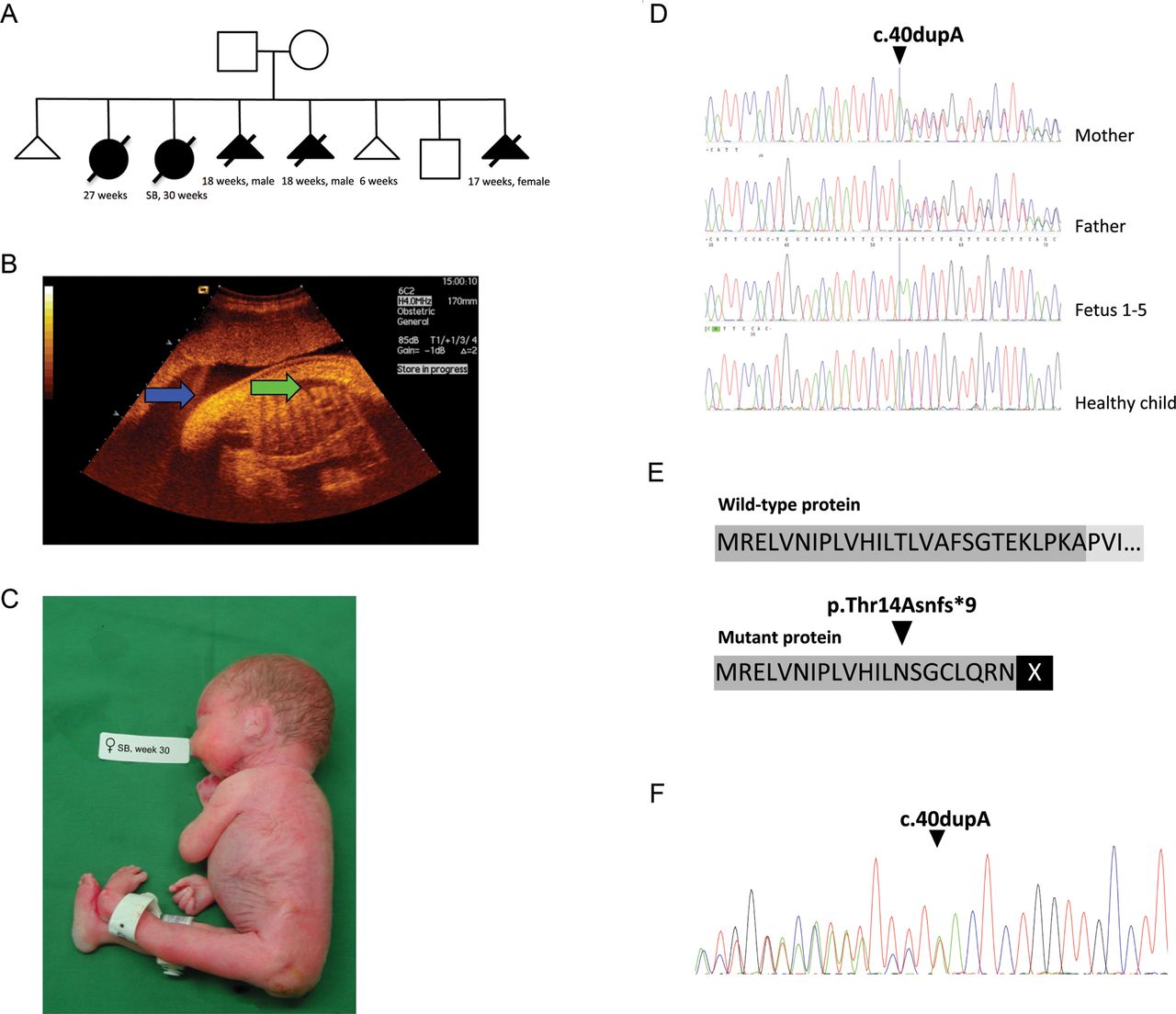
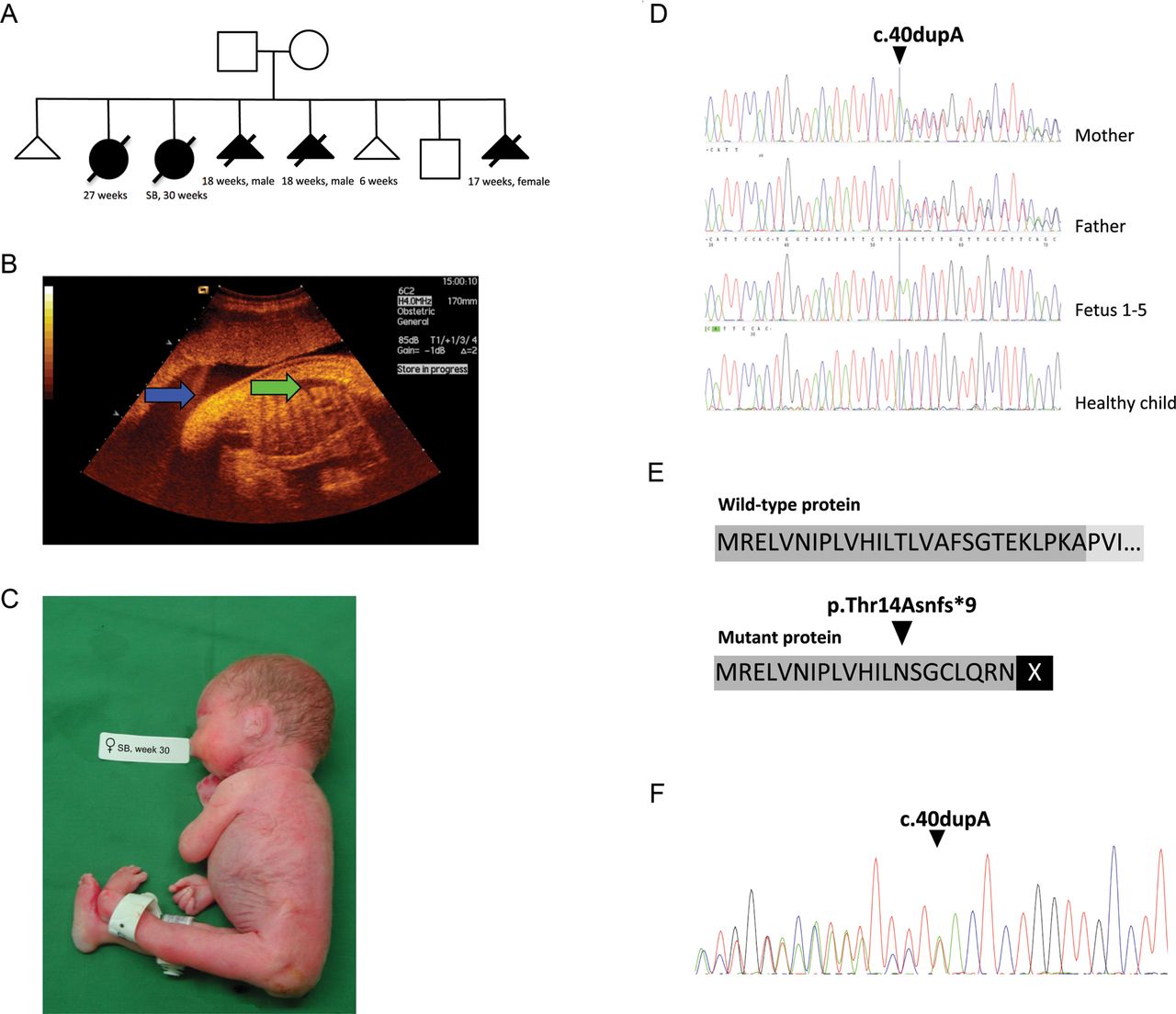
The history of the affected family, showing the pedigree, ultrasound picture and a picture of one affected fetus combined with genetic data revealed a frameshift mutation in muscle, skeletal receptor tyrosine kinase (MuSK) potentially leading to a non-functional protein. (A) Pedigree of affected family displays two miscarriages, five affected children/fetuses, and one healthy child. (B) The second pregnancy displayed polyhydramnion (blue arrow) and fetal hydrops (green arrow). (C) The child with fetal akinesia deformation sequence (FADS) from the second pregnancy was stillborn at week 30. (D) Sanger sequencing revealed that the mother and the father were heterozygous for the insertion, all affected fetuses were homozygous, and the healthy child did not carry the insertion. (E) The insertion causes a frameshift in the open reading frame (ORF) at position p.Thr14Asnfs*9 in exon 1 of MuSK and predicts a premature stopcodon at p.22. (F) mRNA obtained from a muscle biopsy from the heterozygous mother was analysed with primers spanning exon borders (exon 1 and 3) and mRNA molecules containing the insertion was detectable. However, the stop codon at p.22 suggests that a non-functional protein is produced, due to lack of important domains.
Whole-exome sequencing and data analysis
DNA from a trio (mother, father and one affected fetus) was used as starting material for exome sequencing. For each sample, 100 ng of DNA was distributed to 12 separate primer pools and amplified according to the Ion AmpliSeq Exome Library Preparation protocol (Revision A.0, Life Technologies). The separate PCR products were pooled and primer sequences were partially digested. Adaptors were ligated and resulting amplicons were purified using Agencourt AMPure XP Reagent (Beckman Coulter) and eluted in amplification mix (Platinum PCR SuperMix High Fidelity and Library Amplification Primer Mix, Life Technologies). Size-selection and purification was conducted using Agencourt AMPure XP Reagent. The amplicons were quantified using the Agilent Bioanalyzer instrument with Agilent High Sensitivity DNA kit.
Emulsion PCR was performed on the Ion OneTouch 2 system using Ion PI Template OT2 200 Kit v3 chemistry, followed by enrichment using Ion OneTouch ES. Samples were loaded on an Ion PI chip Kit v2 and sequenced on the Ion Proton System using Ion PI Sequencing 200 Kit v3 (200 bp read length, Life Technologies) chemistry. DNA samples for the mother and the father were barcoded and multiplexed on one PI chip, while the fetal DNA was run on a separate PI chip. The sequencing generated over 30 million reads/sample of average length >150 bp.
Alignment of reads to the human hg19 assembly and variant detection was performed using V.4.0 of the Torrent Suite Software. Single nucleotide polymorphism (SNPs) and indel data were stored in a local installation of the CanvasDB in-house database system13 (github.com/UppsalaGenomeCenter/CanvasDB) together with variant annotation information obtained from ANNOVAR14 and dbSNP137. R functions available from the CanvasDB system were used to identify potentially damaging variants that followed a recessive inheritance pattern (heterozygous in the parents and homozygous in the fetus or compound heterozygous in the fetus) and not present (<1.0%) in any of the other 953 exomes stored in the in-house database.
Sanger sequencing
To verify the insertion (c.40dupA, g.113 431 224 dupA) in MuSK (NM_005592.3), genomic DNA were PCR amplified and sequenced in both forward and reverse direction in the parents, all five affected fetuses, and one healthy child. Primers used for PCR and sequencing were forward 5′-GTG GAG CCA TTT TCC TTG CG-3′ and reverse 5′-TTG CCC TAG AAG TCA GAG CTC-3′, creating a 401 bp product. Standard PCR and sequencing conditions were used and are available upon request. Sequence analysis was performed using CodonCode Aligner 5.0.1 software (CodonCode Corporation, http://www.codoncode.com).
RNA extraction, cDNA synthesis and analysis
A muscle biopsy from the mother in the family was prepared and stored in RNAlater (Ambion). The tissue was homogenised and total RNA extracted using the TRIzol method (Invitrogen) according to the manufacturer's instructions. The quality of total RNA was evaluated using Agilent RNA 6000 Nano Kit (Agilent Technologies, St Clara, California, USA), with RNA Integrity Numbers (RIN) of 9.4. cDNA synthesis was performed at 42°C for 60 min using 150 ng of RNA as starting material, 5 µM oligo-dT primer, 5 U RevertAid MuLV transcriptase, 0.8 U RiboLock RNase inhibitor in the reaction buffer supplemented with 5 mM MgCl2 and 1 mM dNTPs. All reagents were from Thermo Scientific. The reaction was terminated by heating for 5 min at 70°C. Primers used for PCR cDNA were forward 5′-AGA GAG CTC GTC AAC ATT CCA-3′ and reverse 5′-ATG CTG TAC CGG GTG TCA A-3′, spanning exon borders and creating a PCR product of 228 bp. Sanger sequencing was performed according to standard conditions and sequence analysis was performed using CodonCode Aligner V.5.0.1 software (CodonCode Corporation, http://www.codoncode.com).
Haplotype analysis
Haploview15 was used to investigate the region for recombination and identify potential founder effect. A standard colour scheme analysis was performed where a logarithm of odds (LOD) <2; D′ <1 (white) shows evidence of recombination and D′=1 (blue) shows strong linkage disequilibrium, which implies little or no evidence of historical recombination. We further investigated 46 blood donor samples originating from the same geographical area in Sweden for the c.40dupA mutation by Sanger sequencing (same conditions as for the previous Sanger sequencing), in an attempt to identify unrelated carriers to estimate the prevalence and age of the mutation.
Muscle biopsies
Haematoxylin and eosin, as well as routine enzyme histochemical staining, was performed on fresh-frozen muscle biopsies from three aborted fetuses at gestational age 17–18 weeks, and from one fetus after the spontaneous intrauterine death. Immunohistochemical analysis was performed on all four biopsies according to standard protocols on 8 µm sections, using mouse monoclonal antibodies towards slow, fast, neonatal and developmental myosin heavy chain (NCL MHCs (1:20 dilution), MHCf (1:10 dilution), MHCn (1:10 dilution), MHCd (1:20 dilution) (Leica Biosystems, Newcastle Upon Tyne, UK)).
Results
Exome sequencing reveals a frameshift mutation in MuSK
WES was performed on genomic DNA from the parents and one affected fetus and 90–92% of the reads mapped to target regions. Average read depth among the samples varied between 83 and 192X. At least 84% of all reads had a target base coverage of 20X. When analysing the data with a recessive inheritance mode in the family trio and filtered towards 953 exomes in the in-house database, only one single variant was identified, a novel insertion of an A in MuSK at position c.40dupA (figure 2D). No compound heterozygous mutations were identified. MuSK (NM_005592.3) is located at position chr9:113 431 051–113 563 278(hg19). This insertion leads to a frameshift in exon 1 of MuSK and predicts a premature stopcodon (p.Thr14Asnfs*9) (figure 2E). The sequence variant was not identified in relevant SNP databases including 6000 exomes (http://evs.gs.washington.edu/EVS/) or in the in-house database with ethnically matched control exomes. Sanger sequencing was performed for verification and segregation analysis of c.40dupA in the family. The mother and father were, as previously shown by WES, heterozygous for the insertion, all affected fetuses were homozygous, while the healthy sibling did not carry the insertion (figure 2D).
Expression analysis of the MuSK c.40dupA allele in muscle
MuSK consists of 15 exons and a protein length of 869 amino acids. To investigate the consequences of the c.40dupA mutation, we analysed RNA extracted from a muscle biopsy derived from the mother. RT-PCR was performed and subsequent Sanger sequencing of cDNA revealed that both the wild-type and the mutated allele were expressed (figure 2F). The results thus indicate that c.40dupA mRNA is not cleared by nonsense-mediated decay (NMD).16 ,17
MuSK insertion is potentially a founder mutation
The affected fetuses were all homozygous for the c.40dupA mutation. To investigate whether the mutation reflects the presence of a mutational hot spot or a common founder, a haplotype analysis of the region was performed. The result revealed a shared haplotype block of 4.02 Mb in size (see online supplementary file; S1) spanning position chr9:111 625 629–115 652 881(hg19). The average recombination rate is 1.1–1.4 cM/Mb for this region in the human genome (based on deCODE).18 A founder effect for this mutation is therefore suggested by the shared 4.02 Mb haplotype on chromosome 9 between the parents and one offspring. We also investigated 46 blood donor samples coming from the same geographical area as where the mutations were identified, but the c.40dupA mutation was not present in any of these samples (data not shown).
Muscle biopsies
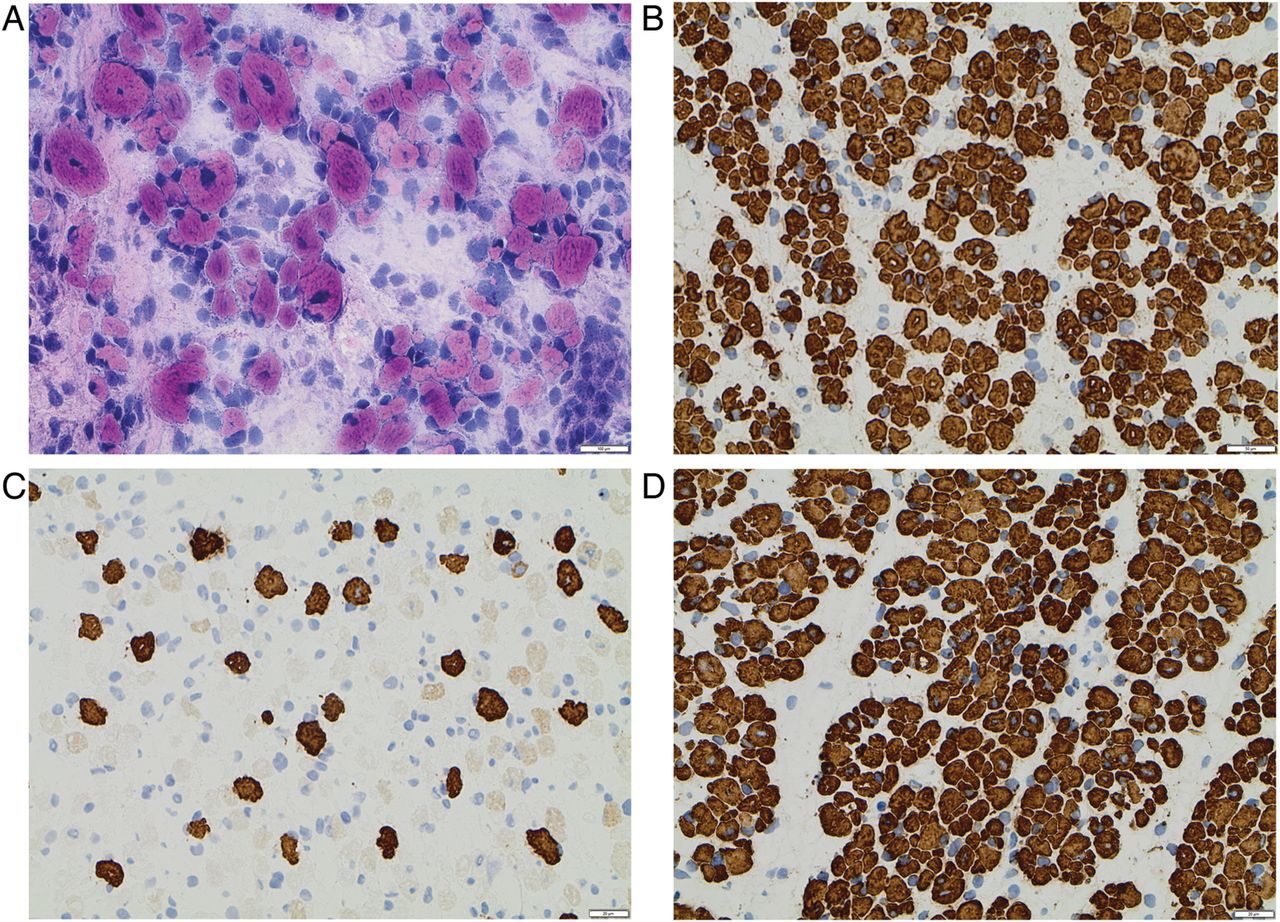
A large variation in fibre size with significant atrophy was demonstrated in all muscle biopsies. Muscle fibre diameter varied from <5 µm in many muscle fibres to a maximum of 15 µm in a few fibres. The muscle fibres co-expressed neonatal and developmental myosin. Only a few scattered fibres measuring 7–12 µm expressed slow myosin. The vast majority of the fibres, including fibres with diameter <5 µm, expressed fast myosin. Increased amounts of loose connective tissue were demonstrated around and within the muscle fascicles (figure 3A–D).
{kind=link}
{kind=link}
{kind=link}
Histological and immunohistochemical findings in the muscle biopsy of one aborted fetus. (A) Haematoxylin and eosin staining showed a large variation in muscle fibre size with many atrophic fibres and increased amounts of loose connective tissue. (B) Immunohistochemistry with antibody towards fast myosin demonstrated that the majority of muscle fibres, including very atrophic fibres, were of type 2. (C) Immunohistochemistry with antibody towards slow myosin demonstrated only a few scattered type 1 fibres, usually of larger diameter. (D) The expression of developmental myosin was identified in all muscle fibres.
Discussion
We report here, for the first time to our knowledge, that a mutation in MuSK is associated with autosomal recessive FADS. MuSK is a muscle-specific receptor tyrosine kinase required for formation and maintenance of the NMJ, a chemical synapse between motor neurons and skeletal muscle fibres. Signalling is achieved by the neurotransmitter acetylcholine (ACh), which binds to the AChR and eventually results in muscle contractions. The development of NMJ is initiated by muscle cells producing and expressing AChR. Agrin is a proteoglycan released by branching motor nerves. It binds to LRP4 and activates MuSK that signals via DOK7 and RAPSN (receptor-associated protein of the synapse) to help stabilise and maintain accumulations of AChR, a process called prepatterning, to develop the NMJ.19 The activation of MuSK requires a dual activation, extracellularly via agrin and intracellularly from DOK7. AChRs are composed of five homologous, membrane-spanning subunits expressed in two different subunit structures: the embryonic-type AChRγ consisting of α2βγδ subunits (predominantly during embryonic development); and AChRε consisting of α2βεδ subunits (replaced in adult muscle).20 MuSK is highly conserved across most species, indicating its essential biological role.
MuSK contains three Ig-like domains, a Frizzled-like cysteine-rich domain, a transmembrane helix, and a cytoplasmic tyrosine kinase domain21 (see online supplementary file; S2). The first two Ig-like domains are important for agrin activation of MuSK. The Frizzled-like domain is important for the ability of MuSK to co-cluster with rapsyn and AChRs.22
Mouse models with MuSKV789M/− demonstrate severe muscle weakness and defects in structure and function of NMJs, showing that mutations in MuSK are the cause of myasthenic syndromes.23 Of note, incomplete loss of MuSK function will cause a CMS phenotype, whereas complete loss of function is lethal. Nine different mutations in MuSK, leading to reduced expression and stability of MuSK and decreased AChR aggregation, have previously been found in autosomal recessive CMS. All of these mutations are located in important domains of the MuSK protein structure (the Ig-like domains, the Frizzled like domain or the cytoplasmic tyrosine kinase domain) (see online supplementary file; S2).24–29
Histological and immunohistochemical findings in the muscle biopsies revealed many atrophic fibres with a predominance of type II fibres and an increased amount of loose connective tissue. This is in contrast to a previous report,27 where compound heterozygous missense mutations in MuSK have been described as causative for CMS and a type I predominance in muscle fibres was described.
In this paper, we demonstrate that complete loss of MuSK is associated with FADS. We propose that the mutation c.40dupA most likely is a founder mutation, due to the long shared haplotype spanning 4.02 Mb between the parents. This haplotype spans more than 20 genes, but the age of the mutation cannot be estimated due to limited numbers of individuals and lack of unrelated carriers included in the analysis. Studies are presently in progress to establish the predicted frequency for the c.40dupA mutation in the Swedish population.
The c.40dupA mutation reported here associated with FADS is located in exon 1 and leads to a frameshift in MuSK predicting a premature stop codon. Transcripts with premature stop codons are frequently targets of nonsense-mediated mRNA decay (NMD). NMD targets newly synthesised mRNA (the spliced product of pre-mRNA) and eliminates abnormal transcripts with premature stop codons, preventing production of truncated proteins that could function in dominant-negative or other deleterious mechanisms.17 However, expression analysis of a muscle biopsy revealed that the mutated allele was expressed in the mother, who was a carrier of the mutation. As a general rule, only premature termination codons located <50–55 bp upstream of the last exon–exon junction manage to escape NMD.30 Nevertheless, there are exceptions to this rule. Recent studies have indicated that LQT2 nonsense mutations near the translational start site of hERG can escape NMD.31 ,32 Considering that MuSK Thr14Asnfs*9 is located close to the initiation codon, one can hypothesise that this transcript also escapes NMD, originating in a short peptide lacking all the important domains of MuSK (the Ig-like domains, the Frizzled like domain, and the cytoplasmic tyrosine kinase domain), which results in a non-functional protein. However, additional functional studies are required to investigate further whether escape from NMD occurs or not.
Interestingly, mice knock-out studies have shown that mice lacking MuSK or agrin show a similar phenotype to the fetuses/children with FADS reported here,33 although the development of NMJ differs in mice and humans. In mice, the switch from embryonic (γ-subunits) to adult (ε-subunits) AChRs takes place in the first 2 weeks of life,34 but in humans the switch occurs earlier and is complete by 31 weeks of gestation. Still, the clinical signs of MuSK deficient mice are similar to humans. Crosses with heterozygous F1 generation showed no clinical signs of disease and the mice developed normally and were fertile. However, F2 generation, homozygous for MuSK deficiency, was lethal. The mice were born alive, but were not able to breathe. They responded poorly upon stimuli and the alveoli air sacs were not expanded. Muscle structure appeared normal, but the NMJ did not form, suggesting a failure in induction of synapse formation.33 Similar knock-out models in mice have been presented for RAPSN35 and DOK7,36 demonstrating essential roles in the neuromuscular synaptogenesis. Knock-out models that lack the embryonic type of AChRγ subunit cause premature death of mice embryos shortly after birth,37 while prevention of expression of adult AChRε is more important during postnatal development of skeletal muscle and the mice live about 3 months after birth.38
The clinical phenotype of the fetuses described here is similar to many previously reported fetuses with mutations involved in the NMJ pathway (table 2). Fetal akinesia and joint contractures have been described in all cases. Distal muscle hypoplasia and facial dysmorphism are present in most cases (except the fetuses with SYNE1 mutations). Pterygium varies, although it is considered a consequence of fetal akinesia. Hygroma, hydrops and pulmonary hypoplasia are more common in mutations in compartments of the AChR, but do also occur in RAPSN and in MuSK as reported here (table 2).6–11
Clinical comparison of FADS syndromes with genes included in the neuromuscular junction pathway
MuSK is located in the same AChR pathway as several other genes reported to cause CMS and/or FADS (CHRNA1, CHRND, CHRNG, RAPSN, DOK7, CNTN1 and SYNE1).6–11 All mutations are homozygous or compound heterozygous mutations. Loss-of-function mutations occurring in CHRNG, CHRNA1, CHRND, CNTN1, DOK7 and MuSK are lethal, whereas missense mutations in RAPSN and splice-site mutation in SYNE1 have been described to cause a less severe phenotype with an outcome that is not always lethal (table 2). RAPSN mutations show contractures and life-threatening respiratory distress6 and SYNE1 mutations cause autosomal recessive arthrogryposis.10
Together with previous findings in knock-out mice models and families reported with MuSK-associated CMS, we propose that whereas incomplete loss of MuSK function will cause a CMS phenotype, complete loss of function will cause FADS. This model of disease pathogenesis has previously been suggested for both DOK7 and RAPSN mutations8 ,9 and now also for MuSK.
Acknowledgments
We wish to thank the family for participating in this study. This study was supported by grants from the Borgström foundation and foundations from Uppsala University Hospital. Ion proton sequencing was performed at the Uppsala node of the National Genomics Infrastructure (NGI).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors All of the authors contributed significantly to this research. M-LB, KEu, GA and MW designed and coordinated the study. KEu and KEr assessed the clinical findings of the cases. AA, MW and SE performed the molecular genetic studies and analysed the data. MW performed the expression analysis studies. OCB and KEr executed the muscle biopsy analysis. MW, JK and ND sampled and genotyped blood donors. MW and M-LB wrote the draft of the manuscript with input from the other co-authors. All authors read and approved the final manuscript.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethics committee, Uppsala University.
-
Provenance and peer review Not commissioned; externally peer reviewed.