Article Text

Abstract
Background Constitutional DICER1 mutations have been associated with pleuropulmonary blastoma, cystic nephroma, Sertoli-Leydig tumours and multinodular goitres, while somatic DICER1 mutations have been reported in additional tumour types. Here we report a novel syndrome termed GLOW, an acronym for its core phenotypic findings, which include Global developmental delay, Lung cysts, Overgrowth and Wilms tumour caused by mutations in the RNase IIIb domain of DICER1.
Methods and results We performed whole exome sequencing on peripheral mononuclear blood cells of an affected proband and identified a de novo missense mutation in the RNase IIIb domain of DICER1. We confirmed an additional de novo missense mutation in the same domain of an unrelated case by Sanger sequencing. These missense mutations in the RNase IIIb domain of DICER1 are suspected to affect one of four metal binding sites located within this domain. Pyrosequencing was used to determine the relative abundance of mutant alleles in various tissue types. The relative mutation abundance is highest in Wilms tumour and unaffected kidney samples when compared with blood, confirming that the mutation is mosaic. Finally, we performed bioinformatic analysis of microRNAs expressed in murine cells carrying specific Dicer1 RNase IIIb domain metal binding site-associated mutations. We have identified a subset of 3p microRNAs that are overexpressed whose target genes are over-represented in mTOR, MAPK and TGF-β signalling pathways.
Conclusions We propose that mutations affecting the metal binding sites of the DICER1 RNase IIIb domain alter the balance of 3p and 5p microRNAs leading to deregulation of these growth signalling pathways, causing a novel human overgrowth syndrome.
Statistics from Altmetric.com
Introduction
Overgrowth, marked by large body size and mass, has numerous aetiologies. Evaluation in the perinatal period assists in distinguishing normal growth variants from endocrine abnormalities and underlying genetic syndromes. Dysmorphic features, developmental delay and asymmetric growth patterns are signs that suggest a genetic aetiology.1 There are numerous phenotypically recognisable overgrowth syndromes, classified by their distinct pattern of features. Prominent among these characteristics are macrocephaly and symmetric overgrowth. Examples of these syndromes include Cowden syndrome (CWS1 (MIM 158350)), Bannayan–Riley–Ruvalcaba syndrome (MIM 153480) and macrocephaly/autism syndrome (MIM 605309), all caused by inactivating mutations in Phosphatase and tensin homolog (PTEN),2 ,3 as well as SOTOS (MIM 117550) and Weaver syndromes (MIM 277590), which are caused by haploinsufficiency of NSD14 and EZH2,5 respectively. Additional phenotypic findings include epidermal changes, cancer predisposition, and distinct facial features, which help to distinguish these syndromes from one another.
Many overgrowth syndromes present with asymmetric overgrowth of bilateral structures, which has been shown to result from mosaic distribution of genetic mutations.3 ,6 The most striking examples of such conditions are Proteus syndrome (MIM 176920), caused by activating mutations in AKT1,7 and congenital lipomatous overgrowth, vascular malformations and epidermal nevi (MIM 612918) syndrome, caused by activating mutations in PI3KCA.8 Additionally, megalencephaly-capillary-malformation-polymicrogyria syndrome (MIM 602501), is caused by mosaic activating mutations in PIK3CA, PIK3R2 and AKT3.9 The mosaic distribution of these mutations has led to the hypothesis that constitutive, gain of function zygotic mutations in these genes are lethal.6 These sporadic genetic conditions eluded molecular analysis until the advent of massively parallel sequencing technologies which allowed identification of de novo mutations. Given the phenotypic overlap among overgrowth syndromes, it is not surprising that many of their molecular aetiologies cluster in pathways that regulate growth. One such pathway is the PIK3–AKT–mTOR signalling pathway,10 in which both activating mutations in drivers of the pathway (AKT1, PI3K) and inactivating mutations in negative regulators (PTEN) have been reported as pathogenic.3 ,4
Syndromes of deregulated growth are often associated with cancer predisposition and specific syndromes are associated with particular tumour types including hepatoblastoma, pheochromocytoma and Wilms tumour.11 Wilms tumour is the most common paediatric renal tumour diagnosed after 1 year of age.12 Most cases are sporadic and without a clear aetiology. However, in some cases it can be associated with specific genetic syndromes, including Beckwith–Wiedeman syndrome (BWS (MIM 130650)).13 ,14
We describe a unique association of congenital nephromegaly, bilateral Wilms tumour, somatic overgrowth, developmental delay, macrocephaly and bilateral cystic lung lesions in the absence of other known genetic aetiologies that defines a novel genetic syndrome.
Materials and methods
DNA extraction
DNA was extracted for each case from peripheral blood mononuclear cells using a QIAGEN QIAamp Blood Mini kit. DNA was also extracted from tumour, bone marrow and unaffected kidney samples using a QIAamp DSP DNA FFPE Tissue Kit.
Whole exome sequencing
Exome sequencing was performed on DNA extracted from peripheral mononuclear blood cells from Case 1 and his unaffected parents, as well as DNA isolated from tumour samples from Case 2 and was consented under an approved IRB protocol. The sequencing library for each sample was prepared using 3 µg of genomic DNA by following the Agilent SureSelect Target Enrichment System for construction of an Illumina paired-end sequencing library (protocol V.1.0.1). The Agilent SureSelect Human All Exon 50Mb Kit was used for the exome capture. Sequencing was performed as 76 bp paired-end reads generated from three lanes of an Illumina GAIIx instrument, generating a total of ∼82 million paired-end reads per sample. Base-calling was performed using the real time analysis software provided by Illumina. The sequence reads were aligned to the human reference genome, human_g1k_v37.fasta, downloaded from the Genome Analysis Toolkit (GATK) resource bundle using Novoalign from the Novocraft Short Read Alignment Package. SAMtools was used to sort the aligned Binary Alignment/Map (BAM) files and potential PCR duplicates were marked using Picard. GATK was used to perform local realignment and base quality recalibration for all three samples. The mean coverage across the protein-coding region of RefSeq genes was 76–80X with approximately 88% of the targeted bases covered by ≥10 reads for each exome. Both single nucleotide variants and insertions and deletions less than 10 bases (INDELs) within the protein coding regions of the RefSeq genes were called using the GATK ‘Unified Genotyper’ tool. Variants resulting in amino acid changes with minor allele frequencies <1% in the population were selected. Further, to identify de novo variants, novel variants that were heterozygous in the proband and homozygous for the reference base in both parents were selected. The exome capture kit provides 100% coverage of all exons within the DICER1 locus, which allowed reliable detection of additional mutations in the tumour sample of Case 2. We determined copy number using the ratio of read number for every exon of DICER1.
PCR and Sanger sequencing
We confirmed the mutation in Case 1 identified by WES by performing PCR on DNA isolated from mononuclear blood cells. Primers flanking the mutation were designed using Primer3 software and run with Invitrogen Platinum Taq. To determine if there were additional ‘second hit’ mutations in DNA isolated from tumour samples, primers published by Hill et al15 were used with the exception of the primers listed below, which were changed due to the presence of polymorphic SNPs within their annealing sites. All PCR products were purified using an Invitrogen Purelink PCR Purification Kit and sequenced using an Applied Biosystems 3730 Capillary DNA Analyser.
PCR primer sequences:
-
Exon 25 F 5′-AAGCTTACGGTTCCACTTCG-3′
-
Exon 25 R 5′-GTGTTGTTGACCAGGGCAGA 3′
-
Exon4F 5′-GGTTCTCTGAAGCGAGTGACAAAA A-3′
-
Exon 4R 5′-GCATCCGGCCTTTATTTGGTTTT-3′
-
Exon 5F 5′-CTTTTCCCATGCTTTCCTGATTTCC-3′
-
Exon 5R 5′-AAGGCTATAACAAGAGAGACATCGCCA-3′
-
Exon 8F 5′-GCTGGGTTACATTTAAGGAGCCATT-3′
-
Exon 8R 5′-AAATCCCAGTTAAACCCCAC-3′
PCR conditions: 95°C for 3 min followed by 40 cycles of 95°C for 30 s, 55°C for 45 s, and 72°C for 1 min 30 s and finally 72°C for 5 min.
Pyrosequencing
Pyrosequencing was performed on DNA isolated from blood (Case 1) or bone marrow as a surrogate (Case 2), tumour samples and unaffected kidney samples, isolated from paraffin blocks, from both cases. Pyrosequencing reactions were performed on all samples in triplicate according to the standard vacuum prep protocol for the PSQ 96 MA instrument from QIAGEN. This method has been validated for the quantification of allele distribution within a heterogeneous population of alleles.16 ,17
Pyro primer sequences:
-
DICER1 Pyro F 5′ GCACAAGCTTACGGTTCCACTTC 3′
-
DICER1 Pyro R 5′ CGCGGGTCTTCATAAAGGTG 3′
-
DICER1 Pyro S 5′ CTTAGAATTCCTGGG 3′
MicroRNA bioinformatics analysis
A list of microRNAs which are differentially expressed in murine cells lines with carrying mutated human DICER1 RNaseIIIb domain ‘hot spots’ was obtained from the supplemental materials of Anglesio et al.18 We selected only 3p microRNAs which show an increase of expression which is greater than 50-fold when compared with mouse embryonic stem cell Dicer −/− controls (see online supplementary table S1). These microRNAs were entered into the DIANA miRPath V.4.0 (updated from V.2.0)19 analysis software (available at http://diana.cslab.ece.ntua.gr/pathways/). We used the cut-off of –ln(p value union) of 20 to identify the pathways which are most significantly shared by these miRNAs.
Results
Subjects
Case 1
The first affected individual presented as a 9-month-old boy with macrosomia and large kidneys. The patient was born via caesarean section at 38 weeks to a 30-year-old G1P1 mother; paternal age was 31 years at birth. Maternal medical problems during the pregnancy included bleeding and spotting. The mother did not take any medications during the pregnancy and there were no known teratogenic exposures. It was noted that the fetus started kicking late in the pregnancy. Prenatal ultrasound at 24 weeks showed enlarged kidneys. Family history was significant for the mother having double ureters on left side, and frequent urinary tract infections. Birth weight was 10 pounds, 13 ounces (>98th centile). Respiratory distress prompted chest imaging which revealed bilateral lung cysts (figure 1A). Renal ultrasound showed multiple small cysts.

Phenotypic features of GLOW syndrome. (A) Coronal view of bilateral lung cysts of Case 1. (B) Case 1 at 9 months of age. MRI shows Wilms tumour in the left kidney (arrow). (C–E) Facial features of Case 1 at 9 months (C) and 28 months of age (D–E) including frontal bossing, a very large anterior fontanelle, hypertelorism, anteverted nares, a flat nasal bridge and slight micrognathia. (F–H) Additional findings in Case 1 at 28 months of age included (F) fat pads on dorsum of foot and toes, (G) rugated soles of feet with skin folds and (H) protuberant abdomen. (I) Coronal views of bilateral lung cysts of Case 2 at the age of 21 months. (J) Case 2 at 13 months of age with Wilms tumour (outline) in the right kidney, with associated ‘claw sign’.
His development milestones were delayed: at 9 months of age, he could not yet roll over and had weak muscle tone. Physical examination at 9 months of age showed a height of 76.5 cm (90th centile), weight of 9.6 kg (75th centile) and head circumference of 52 cm (>98th centile). Additionally, hands were observed with a doughy texture. Hand length was 10.2 cm (>90th centile) and palm length was 6.5 cm (>97th centile). Palmar creases were normal and fingers were tapered. The feet were large and measured 12 cm (75th centile). An abdominal ultrasound confirmed nephromegaly with the left kidney measuring 7.79 cm and the right kidney measuring 7.71 cm. A soft tissue mass within the superior pole of the left kidney was identified that measured 1.95×1.87×1.54 cm (figure 1B). Nephrectomy was performed and histological examination demonstrated the presence of blastemal predominant Wilms tumour confirmed by WT1 staining. The kidney histology was abnormal: numerous immature and abnormally developed glomeruli were present with overall underdevelopment of the renal cortex, in addition to dysplastic medullary ray nodules, abnormal medullae and perilobar nephrogenic rests. The reporting pathologist commented that the histology of the kidney was remarkably similar to that of BWS. However, methylation analysis of 11p15 and genetic testing for mutations in CDKN1C were normal. Karyotype of the tumour was normal and showed 46,XY. Additional genetic work up for macrocephaly-associated genes included mutation analysis for folliculin (FLCN), Nuclear receptor binding SET domain protein 1 (NSD1), and PTEN which were all negative. Finally, chromosomal microarray analysis did not identify any copy number variations; however, small to moderate CNVs cannot be ruled out based on the threshold of this clinical test.
At 18 months of age, he had an MRI of the brain that showed enlarged lateral and third ventricles. At 28 months of age, weight was 15.5 kg (92nd centile), height 95 cm (90th centile) and head circumference 55 cm (>98th centile). There were dysmorphic facial and cranial features including frontal bossing, a large anterior fontanelle, slight hypertelorism, anteverted nares, a flat nasal bridge and slight micrognathia (figure 1C–E), as well as umbilical and left inguinal hernias. At the age of 28 months he presented with dimples on the sides of his ankles. There were fat pads on the dorsum of foot and toes (figure 1F), pronounced plantar creases on both sides (figure 1G) and a large protuberant abdomen (figure 1H). Other dysmorphic physical findings included pectus excavatum, kyphosis and a sacral dimple. At the age of 5, he was diagnosed with Wilms tumour of the contralateral kidney as well as autism.
Case 2
The second affected unrelated individual presented at the age of 14 months with global developmental delay and macrocephaly. Pregnancy was complicated by preeclampsia. Prenatal ultrasounds and laboratory evaluations were normal. Birth weight was 6 pounds, 7 ounces (15th centile). Macrocephaly was noted at birth, but head CT was negative. Family history was significant for a paternal grandmother who had breast cancer at the age of 50 years and skin cancer.
Early in development, global delay was suspected due to delayed rolling at 7 months. He met the following milestones: tracking between 2 and 3 months, reaching between 3 and 4 months, lifting the head between 4 and 5 months, and sitting at 9 months. Global developmental delay was confirmed by 14 months as he was not crawling and not babbling. At 14 months, his growth measurements were weight 13.5 kg (>98th centile), height 81 cm (75th–80th centile) and head circumference 53 cm (>98th centile) demonstrating somatic overgrowth. He had an MRI of the brain that showed mild volume loss, but no signs of hydrocephalus or other brain malformations.
At 18 months of age, he presented with a large firm mass most prominent in the right upper quadrant. CT scan revealed a large mass, arising from the right kidney as well as a multiloculated cystic mass within the upper pole of the left kidney measuring 2.15×2.13×1.84 cm (figure 1J). Left kidney size at this time was 8.1 cm. Right nephrectomy was performed and histological analysis confirmed Wilms tumour. Karyotype for the tumour showed 46,XY. CT of the chest at 21 months demonstrated numerous thin walled cysts throughout the lungs bilaterally (figure 1I). MRI of the brain at the age of 2 showed enlargement of the cerebral spinal fluid (CSF) space in the anterior middle cranial fossa and in the Sylvian fissures bilaterally. At 30 months of age, there was tumour recurrence to the paraspinal area, spleen, contralateral kidney and lungs, which again confirmed to be Wilms’ tumour by biopsy of the paraspinal mass. In addition to macrocephaly, his facial features included a depressed nasal bridge, a left ear pit, frontal bossing and hypertelorism with normal hand and foot creases. He had a karyotype performed on blood, which was normal. A summary of the clinical findings is provided in table 1.
Description of patients in the study
Sequence analysis
Case 1
Whole exome sequencing analysis on DNA isolated from peripheral mononuclear blood cells identified a heterozygous de novo variant in DICER1 (HGA 19g.95560451 A>T, c. 5138 A>T, p.Asp1713Val). The parents shared many rare variants with the proband, confirming biological parentage. Additionally, Sanger sequencing confirmed that this mutation was de novo as it was absent from parental DNA extracted from blood mononuclear cells samples. This variant is not present in dbSNP137, Exome Variant Server Data Release ESP6500SI-V2 or within the UCLA Clinical Genomics Center exome dataset (table 2).
Filtering of variants identified by whole exome sequencing
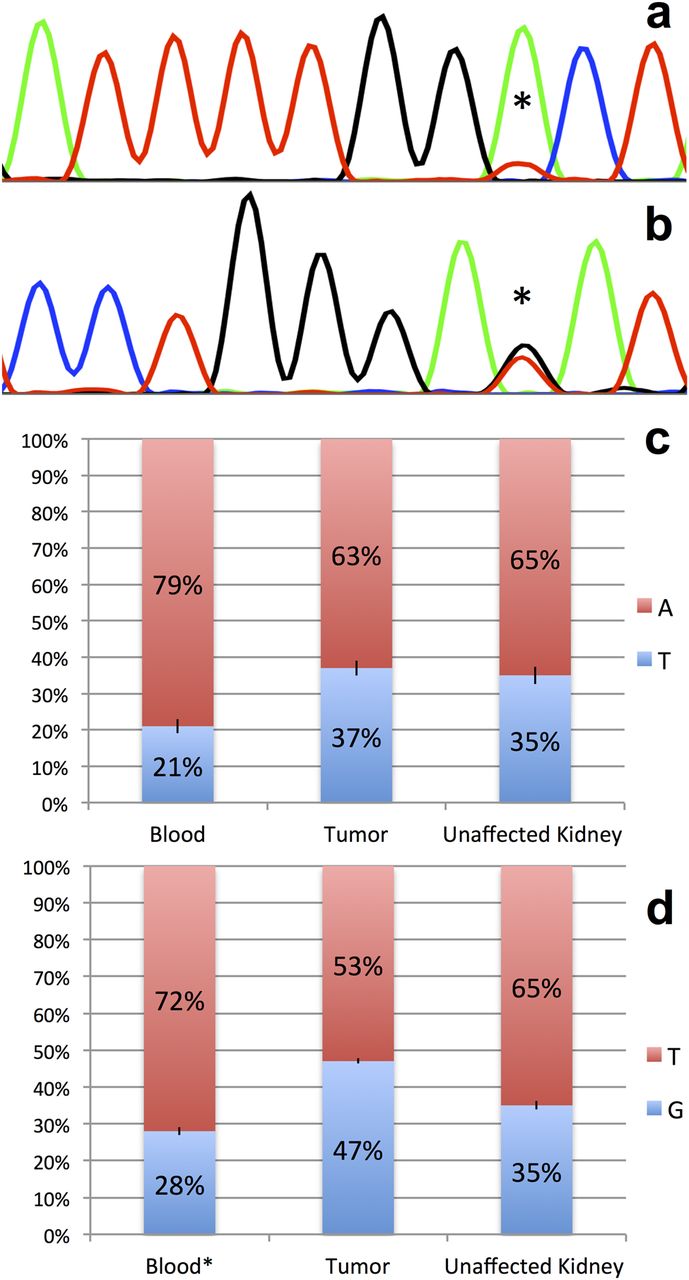
This mutation is ‘probably damaging’ with a score of 1.0 by Polyphen 2.0 software. The mutation was confirmed by PCR and Sanger sequencing. Somatic mosaicism was suspected due to the skewed coverage distribution for the reference allele (48 reads) compared with the alternate allele (10 reads) in the exome sequencing data and the abundance of the peaks observed on Sanger sequencing (figure 2A).

Postzygotic mosaicism of RNase IIIb mutations in DICER1. (A, B) Sanger sequencing revealed a skewed distribution of allele abundance in Wilms tumour samples for each case. (A) Mutation g.95560451 A>T, c. 5138 A>T, p.1713 D>V identified in Case 1 (marked by asterisk). (B) Mutation g.95560438 G>T, c. 5125 G>T, p.1709 E>K identified in Case 2 (marked by asterisk). (C, D) Pyrosequencing identified mosaic distribution of mutations in different tissues. In both cases, percentage of mutation abundance is represented in blue. (C) Case 1 mutation abundance in blood (21%), tumour (37%) and unaffected kidney (35%). (D) Case 2 mutation abundance in blood (28%), tumour (47%) and unaffected kidney (35%). Standard deviations for Pyrosequencing experiments (C,D) are depicted with black bars.
Case 2
DICER1 mutation analysis was performed by Sanger sequencing of the second proband and identified a D1709Y missense mutation (HGA 19, g.95560438 G>T, c. 5125 G>T, p.Asp1709Tyr) located 13 bp from the mutation in Case 1 (figure 2B). This mutation is also probably damaging with a score of 1.0 by Polyphen V.2.0 software.
Mosaic distribution of the mutations
We performed pyrosequencing on DNA extracted from all available tissue samples to determine the relative abundance of the mutations identified in blood, tumour and unaffected kidney samples. This analysis demonstrated mosaic distribution in each tissue, ranging from 21%–37% in Case 1 (figure 2C) to 28%–47% for Case 2 (figure 2D). In both cases, there is relative enrichment of the mutation in both tumour and unaffected kidney when compared with blood.
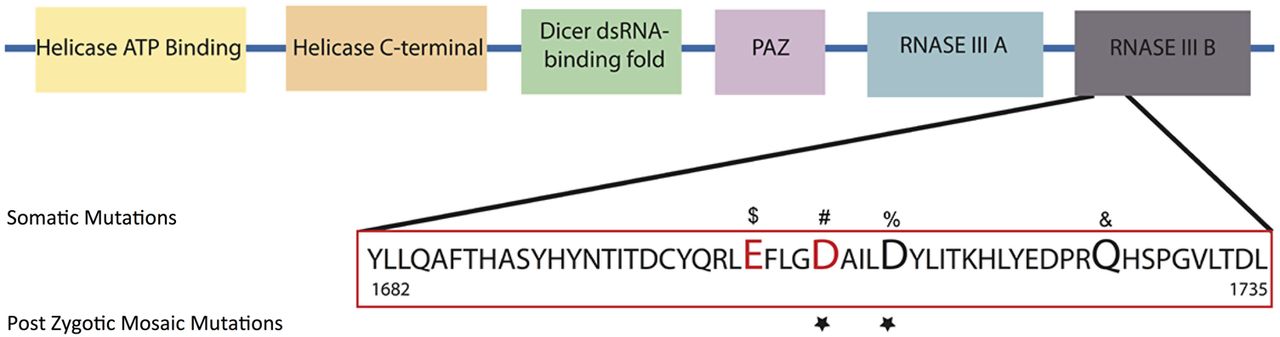
The 1709 and 1713 aspartic acid residues reside in the RNase IIIb domain of DICER1 and are conserved across numerous species.20 Somatic mutations in Arg1709 have been previously reported in a number of cancers (figure 3).21 More specifically, Arg1709 has been identified as part of a metal binding site essential for 5′ microRNA cleavage from mature pre-microRNAs and a hot spot for somatic mutations in cancer.21 Similarly, Arg1713 mutated in Case 1 is located adjacent to this metal binding site, and a case of a somatic mutation in Wilms tumour has been reported (figure 3).22
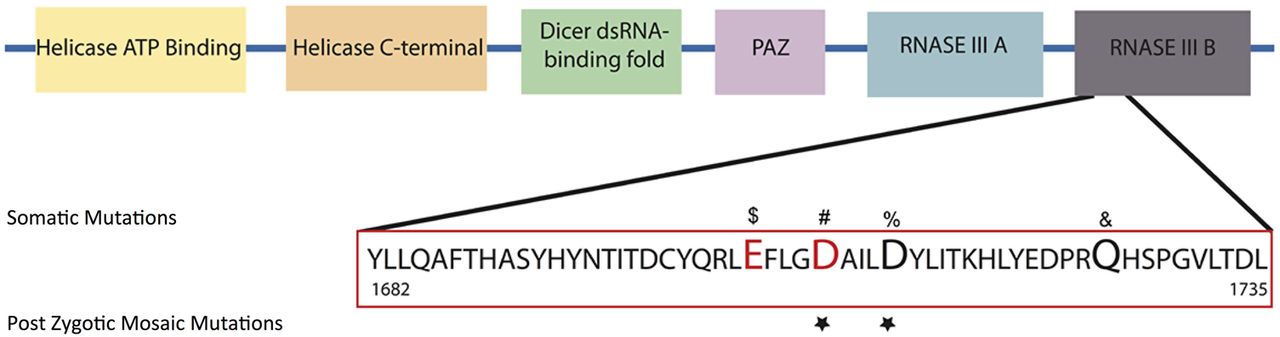
DICER1 RNase IIIb mutations. Schematic representation of DICER1 protein and its domains. Mutations in the RNAase IIIb domain associated with Sertoli-Leydig cell tumours ($, #), non-epithelial ovarian tumour (#), yolk sac tumour (#), juvenile granulosa-cell tumour (#), Wilms tumour (%) and embryonal rhabdomyosarcomas (&) are presented. Residues identified to be cancer ‘hot spots’ and known to reduce 5p cleavage and increase 3p microRNAs according to Anglesio et al18 are depicted in red. Affected residues in GLOW syndrome are marked with a star.
LOH and ‘second hit’ studies
Case 1
There was no evidence of DICER1 copy number loss in the WES data, which was performed on DNA isolated from mononuclear blood cells with all ratios of reads >1. Two additional DICER1 mutations were identified via Sanger sequencing from DNA isolated from tumour, g.95590605C>T, c.1304C>T, p.P453L, located in the Helicase C-Terminal domain (Polyphen 2 Score 0.972: probably damaging) and g.95556912 T>C, c.5529T>C, p.R1898G, located in the double stranded RNA binding domain (Polyphen 2 score 0.969: probably damaging). These mutations are absent from DNA isolated from blood, unaffected kidney as well as parental DNA samples demonstrating that they are tumour-specific de novo somatic mutations. Furthermore, these mutations are unequally distributed within the tumour (see online supplementary figure S1).
Case 2
No evidence of DICER1 copy number loss was identified in the WES data, which was performed on DNA isolated from tumour with all ratios of reads >1. No additional DICER1 mutations were identified by WES.
Bioinformatics analysis
We mined microRNA expression data from Dicer1 null mouse embryonic stem cells transfected with a single copy of human DICER1 affecting the same residue as was seen in Case 2.18 While the phenotype of mice carrying these mutations is not known, we were able to identify a specific population of microRNAs, which are increased in cells carrying the metal binding site mutations (see online supplementary table S1). Additional analysis using DIANA lab mirPath 2.0 allowed us to identify common target pathways shared by these upregulated microRNAs. Our analysis revealed that the following pathways are targeted by 9/10 of the microRNAs: (1) TGF-β (-ln (p value)=24); and (2) MAPK signalling (-ln (p value)=21) and the mTOR pathway is targeted by 8/10 of the microRNAs (-ln (p value)=26) (see online supplementary table S1).
Discussion
DICER1 is a protein that is involved in the microRNA processing pathway.23 Constitutional DICER1 mutations have been associated with cystic lung disease, cystic nephroma, Sertoli-Leydig tumours and multinodular goitres,24 while somatic DICER1 mutations have been reported in additional tumour types.21 ,25–29 Despite the wide-ranging effects of microRNAs on gene expression, the recurrent involvement of specific tissue types including the lungs, kidneys, ovaries and thyroid in cases of DICER1 mutations suggests that tissue-specific microRNAs may play a more prominent role in these organs. The phenotypes associated with DICER1 mutations are varied and reproducible however, to date have not been reported to cause somatic overgrowth, macrocephaly or developmental delay.
The biallelic loss of heterozygosity (LOH) of DICER1 observed in isolated Wilms tumours suggests that DICER1 behaves as a tumour suppressor, requiring a second hit for tumourigenesis to occur.22 Wu et al have reported three cases of isolated Wilms tumour where there is an inherited frame shift deleterious DICER1 mutation in one allele and an acquired mutation in the RNase IIIa (n=1) or RNase IIIb domain (n=2) in the second allele. Case 1 from our report had two second hit mutations in DICER1 which are heterogeneously distributed throughout the Wilms tumour (see online supplementary figure S1). This finding in addition to the absence of second hit mutations or LOH in tumour samples from Case 2 suggests that they may not be necessary or sufficient for tumourigenesis in this syndrome. We cannot rule out that we may have missed mutations in intronic or regulatory regions that may affect DICER1 function. It is also important to note that not all Wilms tumours are associated with DICER1 mutations, as Bahubeshi et al30 have reported a cohort of 50 cases of sporadic Wilms tumours none of which have mutations in DICER1.
Interestingly, of all the mutations reported to date in DICER1, those that alter residues within the RNase IIIb domain are over-represented in sporadic cancers. More specifically, mutations in specific metal binding residues within the RNase IIIb domain are associated with distinct tumour types.18 ,29 We suggest that the phenotypic consequence of these metal binding site mutations is overgrowth and cancer predisposition. These specific DICER1 RNase IIIb mutations act differently than those that cause complete DICER1 loss of function suggesting that DICER1 may also behave as an oncogene.21 ,29 The higher incidence of metal binding site domain mutations in tumours suggests that they can additionally cause overgrowth, macrocephaly and developmental delay when more widely distributed. Similar to P53, we propose that DICER1 can act both as a tumour suppressor as well as an oncogene depending on the specific mutation present and the functional consequence of those changes on protein function.31
The developmental origin of the DICER1 mutation in these cases is likely after zygote formation, which explains their mosaic distribution. It is reasonable to propose that these metal binding site RNase IIIb domain mutations are not tolerated during development and behave differently from haploinsufficient alleles. To support this hypothesis, we have summarised all mutations within the metal binding sites of the RNase IIIb domain of DICER1 and their associated human phenotypes in table 3. As documented in this table, to date there has not been a single case reported of an RNase IIIb metal binding site germline mutation, which in combination with their frequent presence in somatic neoplasms, strengthens the hypothesis that these mutations are not tolerated when inherited.
Reported metal binding site and GLOW syndrome mutations in the RNase IIIb domain of DICER-1 and associated phenotypes
There are four amino acid residues reported to be metal binding sites (1705, 1709, 1810 and 1813) that are essential for RNase IIIb domain function.21 ,29 These metal bindings sites have been labelled as cancer hot spots in DICER1.21 ,29 All are reported to have a similar functional consequence when mutated,18 which is the loss of 5p microRNA synthesis with 3p microRNA synthesis remaining intact. During homeostasis, 5p microRNAs are thought to be the predominant population while 3p microRNAs are normally short-lived and rapidly depleted from cells.18 Case 2 from our study has a postzygotic mosaic mutation that has been reported to be a hot spot at p.Asp1709Tyr, while the mutation in Case 1 affects an amino acid four residues away, p.Asp1713Val. This mutation has been identified in a sporadic case of Wilms tumour.22 We propose that when residue 1713 is mutated it has a similar functional consequence to that of the reported hot spots, causing a similar phenotype as residue 1709 when present in a postzygotic mosaic distribution.
Specific 5p microRNAs have been shown to be essential for the regulation of signalling and growth pathways.32 Furthermore, the balance between specific 3p and 5p microRNAs is essential for the control of growth and prevention of cancer progression.33 This was shown to be the case for mir28-5p and -3p. Using a transwell assay, overexpression of mir28-5p reduces colorectal cancer (CRC) proliferation and invasion in vitro and, conversely, mir28-3p overexpression increases CRC cell migration and invasion in vitro.33 We predict that signalling pathways in which the 5p/3p miRNA balance is essential for correct regulation would be especially sensitive to DICER1 RNase IIIb domain mutations.
We performed a bioinformatics analysis in a murine stem cell model of DICER1 mutation (D1709N) to address our prediction that specific pathways may be affected by a 3p/5p microRNA imbalance. We identified 10 candidate dysregulated 3p microRNAs (see online supplementary table S1). Using DIANA miRPath we generated a list of target genes for each microRNA followed by pathway analysis to identify shared targeted signalling pathways. There were three pathways identified: mTOR signalling, TGF-β and MAPK signalling, all of which were targeted by 80% (8/10) of the 3p microRNAs identified and two of which were targeted by 90% (9/10) of the 3p microRNAs (TGF-B and MAPK). Many of the targets represent negative regulators of each of these pathways, including PTEN, tuberous sclerosis complex (TSC) (mTOR), SKP1a, Smad7, Snuf1 (TGF-β), NF1, Rasa2, Dusp16, Ppm1a and Evi1 (MAPK). These findings suggest that an imbalance in specific 3p microRNAs may lead to excessive cell and tissue growth and cancer predisposition (figure 4). Furthermore, these pathways may represent potential therapeutic targets.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of GLOW syndrome-associated RNase IIIb metal binding site mutations.
We support the hypothesis that specific mutations within the metal binding sites of the RNase IIIb domain of DICER1 allow it to behave as an oncogene, distinct from the effects of frame shift or other deleterious DICER1 mutations.21 The oncogenic microRNA profile manifests as neoplasm formation as well as overgrowth when more widely distributed. We have provided a bioinformatic link between deregulated microRNAs and key growth signalling pathways that provides a potential mechanism by which these mutations may cause tumour formation and overgrowth. While rare cases of nephromegaly34 and renal hamartomas35 have been associated with pulmonary cysts, none have been reported to develop Wilms tumour. We therefore suggest that these domain- and site-specific mutations are causal for a unique constellation of associated clinical features representing a novel syndrome. We have termed this syndrome GLOW for its key findings that include Global development delay, Lung cysts Overgrowth and Wilms tumour. This expands the phenotypic spectrum associated with mutations in the DICER1 gene.
Acknowledgments
We thank the patients and their families. Technical work for sequencing was performed by Traci Lyn Toy. Computational support for sequence data analysis was provided by Bret Harry. Variant annotation was assisted by programmes developed by Michael Yourshaw.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Correction notice This article has been changed since it was published online. Figure 4 has been updated.
-
Contributors SK and JMA performed the experimental, clinical and bioinformatic work and wrote the paper. HL and SFN performed the clinical whole exome sequencing analysis. MAT performed the pathological examination; PK and MI performed clinical evaluation of the cases. SG performed retrospective analysis of all radiographic images.
-
Funding March of Dimes.
-
Competing interests SK is a trainee in the UCLA-Caltech MSTP (NIH T32GM008042) and was additionally supported by a Summer Scholars Grant from the American College of Medical Genetics and the NIH Training Grant in Genomic Analysis and Interpretation (NIH T32HG002536). This work was supported by the David Geffen School of Medicine at UCLA, the Today’s and Tomorrow’s Children Fund, and the March of Dimes Foundation (Grant 6-324).
-
Patient consent Obtained.
-
Ethics approval This study was approved by the Institutional Review Board of the David Geffen School of Medicine at the University of California at Los Angeles.
-
Provenance and peer review Not commissioned; externally peer reviewed.