Article Text

Abstract
Background Knowing the genetic status of patients affected by paragangliomas and pheochromocytomas (PPGL) is important for the guidance of their management and their relatives. Our objective was to improve the diagnostic performances of PPGL genetic testing by next-generation sequencing (NGS).
Methods We developed a custom multigene panel, which includes 17 PPGL genes and is compatible with both germline and tumour DNA screening. The NGS assay was first validated in a retrospective cohort of 201 frozen tumour DNAs and then applied prospectively to 623 DNAs extracted from leucocytes, frozen or paraffin-embedded PPGL tumours.
Results In the retrospective cohort, the sensitivity of the NGS assay was evaluated at 100% for point and indels mutations and 86% for large rearrangements. The mutation rate was re-evaluated from 65% (132/202) to 78% (156/201) after NGS analysis. In the prospective cohort, NGS detected not only germline and somatic mutations but also co-occurring variants and mosaicism. A mutation was identified in 74% of patients for whom both germline and tumour DNA were available.
Conclusion The analysis of 824 DNAs from patients with PPGL demonstrated that NGS assay significantly improves the performances of PPGL genetic testing compared with conventional methods, increasing the rate of identified mutations and identifying rare genetic mechanisms.
- next generation sequencing
- paraganglioma
- pheochromocytoma
- somatic mutations
- mosaicism
Statistics from Altmetric.com
Introduction
Paragangliomas and pheochromocytomas (PPGL) are rare neuroendocrine tumours that can arise either from the adrenal medulla (pheochromocytoma, PCC) or from extra-adrenal paraganglia (paraganglioma, PGL).1 Parasympathetic PGLs mainly occur in the head and neck region (HN-PGLs). In contrast, PCCs and sympathetic PGLs arise from the thoracic, abdominal and pelvic areas (TAP-PGLs). PPGLs are considered as the human tumours with the highest frequency of hereditary cases with at least 35% of inherited forms of the disease.2 Driver mutations can also be identified at somatic level, and overall, germline or somatic mutations in one of the 18 known genes involved in PPGL pathogenesis, generally mutually exclusive, are present in about 60% of tumours.3 These PPGL genes include VHL, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, FH, MAX, EPAS1, HRAS, EGLN1, ATRX, MET, MDH2 and SLC25A11, demonstrating a high degree of heterogeneity in the genetic determinism of these neoplasms.
Published guidelines on PPGL recommend that genetic testing should be considered in all patients with PPGL.4 For years, a sequential strategy prioritising the Sanger genotyping according to clinical feature-driven diagnostic algorithms was generally used. Given the large number of PPGL susceptibility genes and the high mutation rate, the use of next-generation sequencing (NGS) is now indicated as it allows a simultaneous screening of all PPGL genes of interest.2 Our objective was to validate a unique custom PPGL gene panel allowing the sequencing by NGS of the main PPGL genes in germline and tumour DNAs, including DNA extracted from formalin-fixed paraffin-embedded (FFPE) tissues in order to rapidly identify driver mutations in patients and tumours for guiding follow-up, targeted therapies in case of metastatic PPGL and familial genetic counselling.
Methods
Patients
The procedures used for PPGL diagnosis were in accordance with both internal and international clinical practice guidelines.4 5 Diagnosis was confirmed by histology in every case. All patients provided written informed consent for PPGL genetic testing, collection of samples and subsequent analyses. This work is a part of the COMETE TACTIC study (ClinicalTrials.gov Identifier: NCT02672020).
Validation cohort
The validation cohort is a retrospective cohort comprising 202 previously genotyped DNAs extracted from frozen tumours collected by the French ‘Cortico et Médullosurrénale: les Tumeurs Endocrines’ (COMETE) network, for which multiomics analysis was already available and published,6–11 providing from 190 different and consecutive patients. Mutation analysis for RET, VHL, SDHB, SDHC and SDHD genes was performed by direct sequencing of corresponding germline DNA for each patient. When direct sequencing was negative, VHL, SDHB, SDHC and SDHD were also analysed for the presence of large deletions using the Multiplex Ligation-dependent Probe Amplification (MLPA) method (MRC Holland, The Netherlands). Additional Sanger sequencing was partially performed in germline DNAs (MAX, TMEM127, FH, SDHA and SDHAF2 genes) and tumour DNAs (VHL, RET, NF1, MAX, FH, SDHx, EPAS1 and HRAS genes).8 9 11 An SLC25A11 mutation was identified by exome sequencing in one patient whose tumour was included in this cohort.12 Moreover, the genotyping of hotspot mutations in FGFR1 exons 12 and 14 was carried out by Sanger sequencing as this gene was not included in the design of the NGS panel. An FGFR1 hotspot mutation was identified in three tumours (Unpublished data). MAML3-UBTF fusion transcripts were found in four tumours.13
Prospective cohort
The prospective cohort was composed by 578 consecutive patients with PPGL diagnosed in 70 different centres to whom PPGL genetic testing has been proposed. Among the 623 analysed DNAs, 578 were germline DNAs extracted from blood sampling and 45 were tumour DNAs (7 extracted from FFPE tissues and 38 extracted from frozen tissues).
Nucleic acid extraction
Germline DNA was extracted from leucocytes using standard protocols by QIAamp DNA Midi kit (Qiagen) or Chemagic DNA Blood kit in combination with a Chemagic 360 equipment (Perkin Elmer). DNA and RNA from frozen tumour tissues were extracted and purified using an Allprep Kit (Qiagen). DNA extraction from FFPE tumour tissue was performed using the Maxwell 16 instrument (Maxwell 16 FFPE Plus LEV DNA purification kit) (Promega).
Custom PPGL gene panel
The targeted gene panel for PPGL ‘MASTR Plus SDHv2’ was designed using the MASTR Plus technology (Multiplicom, Agilent Technologies) and included VHL, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, FH, MAX, EPAS1, EGLN1, EGLN2, MDH2, ATRX and HRAS genes. All coding exons were encompassed excepted for RET (exons 8 and 10 to 16), EPAS1 (exons 9 and 12) and HRAS (exons 2 to 4). Primers were designed to cover the whole coding sequence and 20bp of intronic flanking regions, allowing for the detection of mutations in splice sites. To face the difficulties to analyse degraded DNA extracted from FFPE samples, the amplicon mean size was designed at 194bp (range 120pb–230bp in length). The ‘MASTR Plus SDHv2’ panel comprises 451 amplicons designed against the human NCBI GRch37/hg19 reference genome assembly. DNA libraries were prepared according to the manufacturer’s recommendations and 2×150 bp paired-end sequenced on MiSeq platform (Illumina, California, USA) using v2 chemistry according to the standard protocol.
The ‘MASTR Plus SDHv2’ panel and bioinformatics pipeline, described below, were first validated on two DNA samples (NA12878 and NA19240) acquired from Coriell Institute (Coriell Cell Repositories, Camden, New Jersey, USA).
Best coverage was obtained for runs of 24 samples. Nine regions of systematic low coverage were found, likely due to high GC contents or repeated sequences, which may affect primer binding. Low coverage affected 0.4% of targeted regions (251/61 346pb). Length of uncovered loci is listed in the online supplementary table S1. All the gaps affecting coding sequence or canonical splice sites were sequenced by the Sanger method.
Supplemental material
Additional information can be found in the online supplementary methods.
Supplemental material
Data analysis
Demultiplexing was performed using MiSeq Reporter (Illumina, California, USA). Alignment and variant calling were performed using SeqNext (JSI Medical Systems) and PolyDiag (Paris Descartes University) software. PolyDiag software used BWA-MEM as a read aligner and Freebayes, Samtools and GATK as variant callers. The 17 genes included in the ‘MASTR Plus SDHv2’ panel were analysed on tumour DNAs. As, in PPGL, ATRX and HRAS genes have been reported as somatically mutated only,14 15 they were not analysed in germline DNAs of patients. The variant analysis was mainly performed using Alamut Visual 2.7 (Interactive Biosoftware) as an interface. Variant classification into five classes16 was based on the framework published by the NGSnPPGL group taking into account multiple criteria including the frequency of the variant in the general population and disease databases, its description in the literature, the variant type, the co-segregation with the disease in families if any, the co-occurrence with known pathogenic mutation, the in silico predictions and the results of functional or supplemental studies.2
Additional information can be found in the online supplementary methods.
Sanger sequencing
Regions with <30X depth of coverage were analysed using Sanger sequencing. The presence of all variants of interest (VOI), corresponding to variants of unknown significance (VUS, class 3), likely pathogenic (class 4) or pathogenic (class 5) variants16 was confirmed using Sanger sequencing except when they were already known for tumours from the retrospective cohort.8–11 Variants identified in tumour DNA were systematically searched for in corresponding germline DNA when it was available.
Search for large rearrangements
In the prospective cohort, VHL, SDHA, SDHAF2, SDHB, SDHC, SDHD and MAX genes were also analysed for the presence of large deletions or duplications using the MLPA method (MRC Holland, The Netherlands).
Immunohistochemistry
To assess the functionality of some VUS, immunostaining was performed when possible. Paraffin blocks were cut and sections (4–6 µm thick) were mounted on Superfrost Plus slides and used for immunohistochemistry as described previously.17–19 The antibodies used were anti-SDHB (HPA002868, Sigma-Aldrich, 1/500), anti-SDHA (Abcam, ab14715, 1/1000) and anti-2SC.
Droplet digital PCR
Sample partitioning was performed using the QX200 Droplet Generator (Bio-Rad), PCR amplification using the C1000 Thermal Cycler (Bio-Rad) and droplet reading using the Droplet Reader (QX 200), which provides absolute quantification in digital form. TaqMan probe-based assays (Bio-Rad) were used. Each mutated event was detected with a primer labelled with FAM fluorophore and the corresponding wild-type allele with a primer labelled with HEX fluorophore. Reference of Droplet Digital PCR (ddPCR) Mutation Assay (Bio-Rad) for the SDHB c.557G>A nucleotide variation is the following: dHsaMDS127410807.
Results
Validation of the NGS ‘MASTR Plus SDHv2’ panel in the retrospective cohort
To assess the diagnostic accuracy of the ‘MASTR Plus SDHv2’ panel, we first searched for the known mutations previously identified in the validation cohort. Tumour DNA was available for 201 out of the 202 samples of the COMETE collection (online supplementary table S2). Using Sanger sequencing, we had previously identified 119 point or short indel mutations in 116 tumour DNAs (66 mutations at the germline and 53 at the tumour level). All mutations were confirmed by NGS using the ‘MASTR Plus SDHv2’ panel. A germline NF1 pathogenic mutation was found in the 10 patients with a clinical diagnosis of neurofibromatosis type 1 (NF1), but who had not benefited from a previous NF1 molecular testing (online supplementary table S2). Six out of the eight (75%) MLPA-confirmed gross deletions were detected by the NGS assay (two whole VHL deletions, one whole MAX deletion, one SDHC exon 3 deletion, one SDHB exon 1 deletion and one exon 3 VHL deletion). An SDHC exon 2 deletion was not detected due to the absence of coverage of this exon by the NGS panel and a somatic NF1 whole deletion was missed.
Supplemental material
Improvement of mutation detection in the retrospective cohort
Besides the confirmation of the already known mutations, 32 additional VOI were identified using the NGS panel, including unknown VOI in tumours previously considered as ‘negative’ or additional VOI in mutated-tumours (online supplementary figure S1).
Supplemental material
Identification of VOI in ‘negative’ tumours
A variant of interest was identified in 24 of the 70 tumours previously considered as ‘negative’ (ie, without mutation identified by former Sanger sequencing). A likely causative mutation was found through the ‘MASTR Plus SDHv2’ panel in 16 tumours and using the FGFR1 Sanger sequencing in 2 tumours. Class 3 to 5 variants were identified in VHL (n=4), EPAS1 (n=4), RET (n=4), NF1 (n=3), FGFR1 (n=2) and HRAS (n=1) genes. All but one (NF1, p.Leu2650Ser) were present in the tumour DNA only. Moreover, two ATRX likely pathogenic variants (c.242+2T>A and c.5114_5122del) were found in two metastatic tumours, while one ATRX missense variant c.6869A>G, p.Asn2290Ser was found in a benign tumour at both germline and somatic level. In addition, UBTF-MAML3 fusions were identified in four tumours.13
Identification of additional VOI in mutated tumours
Thirteen additional mutations or VUS were detected using the NGS panel and 1 FGFR1 mutation using Sanger sequencing. Four somatic mutations (three in SDHB-mutated tumours and one in a tumour with biallelic FH mutations, all metastatic cases) and one missense VUS (in a benign VHL-related case) were identified in the ATRX gene. One VUS in EGLN1 (p.Ser75Phe) was detected as being associated with a somatic VHL mutation. One somatic FGFR1 mutation was found in a PCC of a patient presenting a missense NF1 germline variant, p.Ala2511Val described as likely benign. Its heterozygosity in the tumour DNA and the occurrence of the somatic FGFR1 mutation are two additional criteria in favour of the non-pathogenicity of the NF1 p.Ala2511Val variation. One PCC, carrying a somatic c.25del, p.Val9Trpfs*56 mutation in the MAX gene with an allelic frequency of 11%, presented an additional c.182A>G, p.Gln61Arg mutation in HRAS, with a higher ratio (35%) revealing a genetic heterogeneity in this tumour. The NGS assay discovered a somatic double hit, corresponding to two different somatic mutations in the same gene, in three tumours (two with NF1 mutations and one with VHL mutations). Finally, three VUS in SDHA were found (two in RET-mutated tumours and one in an SDHB-mutated tumour) with an allelic ratio close to 50%, suggesting that these variants were likely heterozygous. Their association with a driver mutation, their report in the gnomAD database and their non-association with an SDHA second hit in tumours are not in favour of their pathogenicity.
Diagnostic performances of the NGS assay in the prospective cohort
Genetic testing of germline DNA
The NGS ‘MASTR Plus SDHv2’ panel combined with MLPA assays was performed in germline DNA of 541 patients diagnosed with PPGL and identified 120 VOI belonging to class 3 (n=39), class 4 (n=24) and class 5 (n=57) (online supplementary table S3). The most frequent germline mutated genes were SDHB (n=30; 5.5% of patients), SDHA (n=19; 3.5% of patients), SDHD (n=13; 2.4% of patients) and VHL (n=11; 2% of patients) (figure 1). The overall germline mutation rate was of 22% (120/541) when considering all the VOI and 15% (81/541) when considering variants from classes 4 and 5 only. Unfortunately, we were not able to retrieve any samples to study the effect of synonymous variants on mRNA splicing.
Supplemental material

Distribution of VOI in PPGL susceptibility genes identified by NGS panel in germline DNA of 541 patients. NGS, next-generation sequencing; PPGL, pheochromocytoma and paraganglioma; VOI, variants of interest.
Interestingly, the NGS assay allowed identifying three patients presented two germline VOI in two different genes. One patient with a PCC diagnosed at 55 years age carried two missense variants, one in FH (c.151C>T, p.Arg51Trp) and one in EPAS1 (c.1635C>G, p.Ile545Met). Moreover, one SDHB mutation (c.435del, p.Phe146Serfs*12) associated with an SDHD variant of unknown significance (c.158C>T, p.Pro53Leu) was found in a patient with an HN-PGL diagnosed at 26 years age. Finally, two previously described mutations, one in NF1 (c.2033dup, p.Ile679Aspfs*21) and one in SDHD (c.170–1G>T) were identified in a patient who developed both a PCC and a carotid PGL.
Combination of NGS in both germline and tumour DNA
Thirty-seven patients were investigated by NGS assay in both germline (n=37) and tumour (n=45) DNA. VOI were found in 26/37 patients (70%) (online supplementary table S3). Three patients had an SDHA germline mutation. One of them, the intronic deletion c.771–21_771-13del, was heterozygous at the germline level (50% of reads) and compatible with a loss of heterozygosity (LOH) in the tumour DNA (85% of reads). The analysis of the corresponding cDNA extracted from leucocytes showed aberrant splicing (exon 7 skipping) demonstrating the pathogenicity of the SDHA mutation. In the two other cases, both germline and somatic SDHA mutations were identified, leading to a biallelic SDHA inactivation in the tumours. The first tumour associated the germline c.1775A>G, p.His592Arg and the somatic c.1753C>T, p.Arg585Trp SDHA mutations and the second one, the germline c.16663+3G>C and the somatic c.1765C>T, p.Arg589Trp. All these three tumours exhibited negative SDHA and SDHB immunostaining confirming the deleterious effect of these mutations.20 One patient presented a germline missense VUS in EGLN2, c.547G>A, p.Val183Met associated with loss of the wild-type allele (allelic ratio of 48% in the germline and 63% in the tumour DNA). Another patient with a personal and family history of NF1 was found to be a carrier of a germline missense NF1 mutation c.2903T>G, p.Met968Arg also associated with loss of the normal allele (allelic ratio of 48% in the germline and 81% in the tumour DNA). We also identified a hotspot mutation in EPAS1 (p.Pro531Arg) in association with an SDHD c.158C>T, p.Pro53Leu variant at the germline level. In this tumour, SDHB immunostaining was negative, but there was no evidence for LOH at the SDHD locus. Hence, the causative role of each variant in the determinism of this tumour could not be fully determined.
Among tumours with somatic mutations only, one presented biallelic FH mutations ([c.364_367del(;)c.1178C>T]). Immunohistochemistry for 2-succinocysteine (2-SC) performed on the tumour tissue showed positive staining which is in favour of the pathogenicity of both variants. In a patient presenting with metastatic multiple PGLs but no germline mutation, we analysed two different tumours (one PCC and one PGL of Zuckerkandl organ). We identified two different EPAS1 heterozygous hotspot mutations: one tumour carried the c.1591C>T, p.Pro531Ser and the second one the c.1589C>T, p.Ala530Val mutation. Among other tumours, a single somatic mutation was found in VHL (n=7), HRAS (n=4) and EPAS1 (n=3). One heterozygous SDHB missense somatic variation c.563T>C, p.Leu188Pro was discovered in a metastatic PCC harbouring a negative SDHB and a positive SDHA immunostaining, which brought evidence for the pathogenicity of the mutation. One SDHD frameshift somatic mutation c.284_285insAGGG was detected in a patient with multiple HN-PGLs and lymph node metastases. The allelic ratio of the mutation was 27% in the primary tumour and 35% in the metastasis. Mosaicism was excluded in both cases (allelic ratio for the mutated allele in germline DNA was respectively 0/2205 and 0/1703).
Finally, the NGS ‘MASTR Plus SDHv2’ panel allowed identifying two mosaic mutations. The SDHB c.557G>A, p.Cys186Tyr mutation (allelic ratio=42%) was found in the DNA extracted from a TAP-PGL diagnosed at 24 years age (figure 2A). The search for the mutation in the corresponding germline DNA with the NGS assay revealed the presence of the mutation in 15% of the reads, demonstrating the constitutional mosaicism of the SDHB mutation. The presence of the SDHB mutation at the mosaic state was confirmed by Sanger sequencing (figure 2B) and droplet digital PCR (figure 2C). Nevertheless, on the electropherogram of Sanger sequencing, the presence of the mutation in the blood DNA would probably have been missed because the peak corresponding to the mutation could be misleadingly considered as an artefact. In a patient who presented a left PCC at 13 years and a right PCC diagnosed 10 years later, a pathogenic VHL mutation c.482G>A, p.Arg161Gln was found in the DNA extracted from the left PCC (allelic ratio=24%) and the right PCC (allelic ratio=18%). In the DNA extracted from leucocytes, the mutation was found in 6 out of 1898 reads (0.3%), which was difficult to distinguish from the background noise. Droplet digital PCR experiments revealed 26% of mutated allele in the left PCC, 19% in the right PCC and 0.6% in the blood DNA versus 0.09% in the control DNA (data not shown). These findings are in favour of a low level of germline mosaicism, but the analysis of another tissue (ie, buccal cells) would be helpful to confirm it. The low allelic ratio of mutations in tumour tissues could be explained by the high vascular density of VHL-related and SDHx-related tumours leading to high level of contamination by non-mutated cells. In this context of allelic fraction <50%, mutations being secondary genetic events rather than primary driver mutations could not be excluded, but their presence in both tumour and germline DNA confirmed the mosaicism.

Identification of the c.557G>A SDHB mutation at mosaic state. (A) NGS results (antisens). (B) Electropherogram from Sanger sequencing of SDHB exon 6. (C) Quantification of each allele by ddPCR. ddPCR, droplet digital PCR; Mut.Al, mutated allele, n.d, not detected; NGS, next-generation sequencing.
Altogether, the use of the NGS assay in the prospective cohort allowed finding mutations at germline, somatic and mosaic levels (online supplementary table S3). All variants reported by NGS were validated by Sanger sequencing, revealing no false positive. The NGS panel also accurately detected 10 large deletions, which were all confirmed by MLPA.
Discussion
Our results demonstrate that the NGS assay with the custom-designed targeted ‘MASTR Plus SDHv2’ panel significantly improves the diagnostic accuracy for mutation detection in PPGL genes compared with former technical approaches. Several studies previously reported the use of targeted NGS assays for PPGL genetic testing, including various genes.21–24 Recently, the NGSnPPGL study group adopted a modified version of ClinVar’s ‘gold star’ scale to assign an evidence level to each gene reported as implicated in the genetics of PPGL.2 Level 3 and 4 genes comprised those with established evidence or functional validation of involvement in PPGL tumourigenesis at the germline or somatic level. The advantage of our assay, compared with others previously published, is to combine a comprehensive gene panel that includes all the level 3 and 4 genes defined in the consensus statement on PPGL2 to an optimised design allowing analysing germline, frozen tumour DNA and DNA from FFPE tumour tissues in a single assay. The sensitivity of the panel was assessed on 119 point mutations or short indels previously found by Sanger sequencing in the COMETE cohort and reached 100% (online supplementary table S2). Our pipeline was able to easily detect indels, including a 37 bp deletion in the NF1 gene. Considering large rearrangements affecting exons with good coverage, the sensitivity was 86% (6/7). The only gross deletion missed involved the whole NF1 gene and was the unique somatic large deletion found in the cohort. This lack of detection was likely due to important contamination of the analysed tumour tissue by normal cells.
Altogether, the mutation rate in the validation cohort, previously assessed to 65% (132/202) by Sanger sequencing, was re-evaluated at 78% (156/201) after NGS assay analysis, FGFR1 Sanger sequencing and MAML3-UBTF fusion transcript search for (online supplementary figure S1). Among variants identified by NGS, seven had an allelic ratio <20% explaining why Sanger sequencing could miss them. The other ones were found either in genes not previously genotyped by Sanger (ie, ATRX) or because previous genotyping was done partially only (ie, genotyping limited to tumours presenting a specific transcriptomic or LOH profile). It is noteworthy that some somatic events with low allelic fraction may not be pathogenically relevant especially when they do not affect clearly established PPGL susceptibility genes. Interestingly, the new genetic data generated by NGS clarify the classification of the PPGL COMETE collection after transcriptomic analysis.8 9 All tumours classified into the cluster C1B, which was defined as the VHL-related cluster, were explained by either a VHL (26 germlines and 15 somatic) or an EPAS1 mutation (1 case). In the tumours classified in the cluster C2A (defined as the RET/NF1 cluster), mutations were identified in 64/78 (82%), including 36 in NF1, 16 in RET, 7 in HRAS, 3 in FGFR1 and 1 in TMEM127 and MAX. In the cluster 2B-related tumours, four somatic EPAS1 mutations and four MAML3-UBTF fusion transcripts were detected.
Considering the prospective cohort, VOI have been found in all except one (SDHAF2) gene belonging to the ‘MASTR Plus SDHv2’ panel validating the accuracy of the selected gene list (figure 1). Given that the mutation rate is of 15% (class 4 and 5 variants) in patients with an apparently sporadic presentation of the disease (ie, a single benign tumour without any history of the disease or syndromic presentation, 406/541) in our prospective cohort, our data are in accordance with previous publications reporting a mutation rate of 11%–13% in patients with sporadic PPGL.24 25 In patients with a family history of PPGL (n=9), a mutation was found in 100% of cases (four in VHL, two in SDHD, one in SDHB, one in SDHC and one in MAX gene). In all six patients with personal or familial clinical presentation evocating NF1, the causative NF1 mutation was identified. In patients with multiple or recurrent PGLs (n=21), germline mutations were found in 12 patients (57%), all in SDHx genes and interestingly, affected each SDHx gene (SDHA, SDHB, SDHC and SDHD). In patients with a single HN-PGL, mutations were mainly found in SDHx genes (10 in SDHA, 10 in SDHB, 6 in SDHC and 8 in SDHD) but class 3 variants were also found in EGLN2, FH and TMEM127, suggesting their potential role in the development of parasympathetic PGLs and justifying their genotyping in patients with HN-PGL only.
A remarkable finding was the identification of two germline pathogenic mutations, affecting SDHD and NF1, in a patient who developed multiple tumours (PCC and carotid PGL). The SDHD (c.170–1G>T) mutation was previously reported as leading to exon 3 skipping26 and responsible for familial PPGL.26–29 The NF1 (p.Ile679Aspfs*21) mutation was also reported several times in patients with neurofibromatosis type 1.30 The retrospective clinical examination of the patient did not reveal any clinical signs of NF1, which is in accordance with the recent description of pathogenic NF1 mutations in patients with PPGL unsuspicious for NF1.31 For this patient, tumour tissue analyses showed a loss of SDHB immunostaining, a LOH at the SDHD locus and, surprisingly, the loss of the NF1 allele carrying the NF1 mutation. All these data support the role of the SDHD mutation in the development of patient’s tumours and lead to consider the NF1 mutation as an incidental finding, without direct consequence in the current patient’s disease but suggesting that the patient should be screened for NF1 disease. Consequently, presymptomatic testing should be offered to first-degree relatives for both mutations.
Regarding the patient carrying two missense germline variants, one in FH (c.151C>T, p.Arg51Trp) and one in EPAS1 (c.1635C>G, p.Ile545Met), the PCC tissue analysis revealed a LOH at the FH locus and positive 2-SC immunostaining, which are both in favour of the FH, p.Arg51Trp pathogenicity that can be, then, classified as pathogenic. No additional support was provided regarding the EPAS1, p.Ile545Met which is classified as VUS.
No surgery was performed for the patient associating an SDHB mutation (c.435del, p.Phe146Serfs*12) and an SDHD variant of unknown significance (c.158C>T, p.Pro53Leu) preventing tumour analysis. However, the association of SDHD p.Pro53Leu variant with another pathogenic SDHx mutation was already described,32 and experimental studies in yeast have shown that this variant does not alter protein function33 suggesting that SDHD p.Pro53Leu could be a rare non-functional polymorphism.
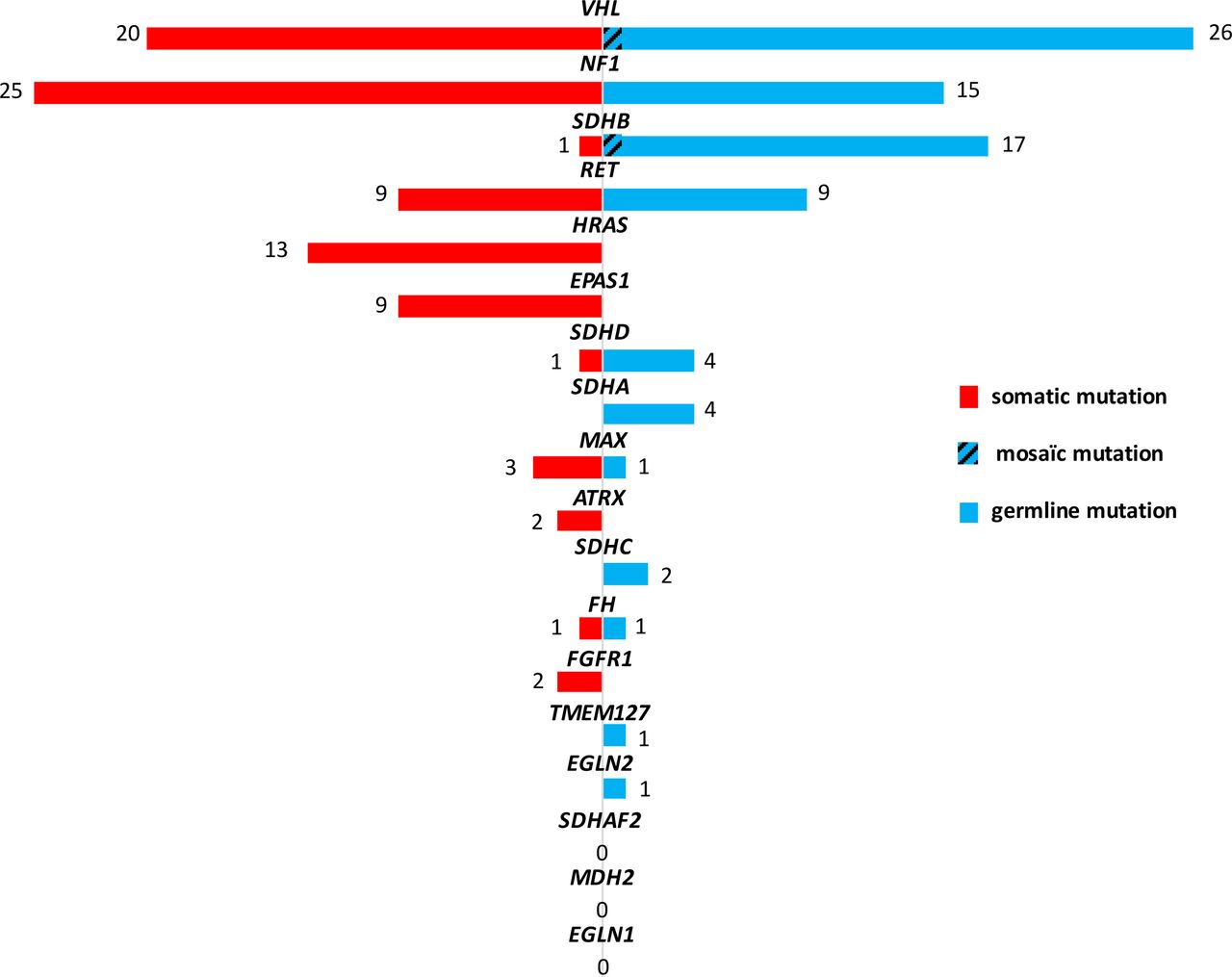
Combining the prospective and the retrospective cohorts, we analysed both germline and tumour DNAs in 227 cases. Among them, 168 (74%) carried a driver mutation: 80 germline (36%) and 86 somatic (37%). The most frequent germline mutated genes were VHL (n=25), SDHB (n=16), NF1 (n=15) and RET (n=9), whereas somatic mutations were mainly found in NF1 (n=25), VHL (n=20), HRAS (n=13), RET (n=9) and EPAS1 (n=9) (figure 3).

![[jmedgenet-2018-105714supp004.jpg]](https://jmg.bmj.com/content/jmedgenet/56/8/513/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
Distribution of VOI in PPGL susceptibility genes identified by NGS panel in 227 patients for whom both germline and tumour DNAs were analysed. NGS, next-generation sequencing; PPGL, pheochromocytoma and paraganglioma; VOI, variants of interest.
Somatic VHL mutations were identified in 13/190 (6.8%) cases of the retrospective cohort and in 7/37 (19%) cases of the prospective cohort. This ratio of somatic mutations is higher than in the TCGA cohort (1.7%)34 but comparable to the one reported by Curras-Freixes et al (7%).24 Nevertheless, it is worth noting that this proportion is probably slightly overestimated as the prospective cohort analysed at the somatic level was enriched in suspicious cases (young age of onset, multiple locations…).
ATRX variants were found in 3.5% of cases (8/227). Among them, seven were identified at somatic level including one in benign VHL-related tumour and six in metastatic cases (three SDHB-mutated, one FH-mutated and two non-mutated tumours13), which is in accordance with the described role of ATRX in the development of aggressive PPGL.13 15 Interestingly, ATRX somatic variants were already reported in benign VHL tumour.15 35 Among the malignant tumours analysed at both germline and somatic level, 18% (6/33) carried an ATRX variant which is slightly lower than the previously reported prevalence (3/12, 25%).34
Interestingly, the patient presenting two different somatic EPAS1 mutations in two different tumours was diagnosed with a heterozygous haemoglobin C disease (haemoglobin C=32%, haemoglobin A2=2.9%). Although this trait has no clinical or biological expression, it could lead to a chronic hypoxic status driving chromaffin cells more sensitive to EPAS1 activating mutations and promoting tumourigenesis, as previously described in patients with cyanotic congenital heart disease.36
Remarkably, this approach identified two SDHx somatic mutations (one in SDHB and one in SDHD), which were rarely reported.37 38 Of note, these SDHx somatic mutations were both present in patients with metastatic PPGL.
This NGS assay was able to detect two mosaic mutations, one in SDHB and one in the VHL gene, in patients with young age of onset. To our knowledge, this is the first report of an SDHB mosaic mutation. VHL mosaicism is better described but usually found in patients with typical VHL disease-associated syndromic lesions.39 40 In the present case, the patient was diagnosed with bilateral PCC only. The high suspicion for germline mosaicism justifies a complete clinical screening for VHL disease. Our results suggest that mosaicism can occur in patients with PPGL (1% of cases in this cohort), and not only in VHL or EPAS1 genes. It was probably underestimated until now due to the low sensitivity of classical sequencing techniques. The current spread of NGS technology use should allow evaluating its prevalence in patients with PPGL. This study demonstrates that deep coverage NGS, combined with ddPCR, allows to discriminate mosaic mutations in patients with PPGL, which is of major importance for family counselling and risk assessment for disease recurrence.
In total, the NGS ‘MASTR Plus SDHv2’ panel assessed in both retrospective and prospective cohorts, encompassing 768 patients’ germline and/or tumour DNA, clearly showed higher accuracy for PPGL genetic testing than conventional methods with a significant increase of the mutation rate. Furthermore, the NGS panel brought an additional value in diagnosing co-occurring variants and mosaicism, both genetic mechanisms not easily identified so far.
The mutational data are essential for guiding genetic counselling, which indicates or exempts (in the presence or the absence of a germline mutation) specific management and follow-up as well as predictive genetic testing in the relatives or to reassure (in front of truly somatic mutations) patients about the risk of relapse.
Finally, the recent knowledge delivered by -omics studies in PPGL molecular pathogenesis gave evidence that patients with metastatic PPGL would benefit from personalised medicine,3 but a precise molecular classification of tumours will be mandatory for guiding targeted therapies. For this purpose, the custom-designed ‘MASTR Plus SDHv2’ targeted panel would be a relevant molecular tool to quickly assess the molecular portrait of tumours by NGS, even when FFPE-DNA is only available.
Acknowledgments
The authors are grateful from Caroline Travers, Valentin Adamus, Nelly Le Pottier, Mathieu Madelaine and Michel Crepin for their technical contribution to this work. They thank Karine Auribault, Patrick Nitschke, Annabelle Venisse, Jurgen Del Favero, Luc Seghers and Hanaa Aissaoui for their help in the development of the NGS assay and the bioinformatics pipeline. They also thank Christophe Simian, Ivana Stankovic, Julien Vidil for technical assistance and Catherine Tritscher, Isabelle du Plessis d’Argentré for administrative assistance. They specifically thank the Biological Resources Center and Tumor Bank Platform, Hôpital Européen Georges Pompidou (BB-0033-00063). They thank the pathologists who provided tumor tissues: Prof Cécile Badoual and Dr Tchao Meatchi (AP-HP, Hôpital Européen Gorges Pompidou, Paris), Dr Mathilde Sibony (AP-HP, Hôpital Cochin, Paris), Dr Abir Ghuzlan (Institut Gustave Roussy, Villejuif), Dr Frédérique Tissier (AP-HP, Hôpital de la Pitié Salpétrière, Paris), Dr Felix Lerintu (Hôpitaux Civils de Colmar), Dr Sophie Michalak (CHU Angers), Dr Valérie Coste-Martineau (Hôpital Arnaud de Villeneuve, CHU Montpellier) and Dr Yves Allory (AP-HP, Hôpital Henri Mondor). They are grateful to the following clinicians and investigators of the COMETE-TACTIC consortium for their support: Delphine Hourton, Stéphanie Chambaud (Clinical research unit), Anne-Marie Madjalian, Anne-Laure Faucon (Hypertension), Franck Zinzindohoue (Surgery), Benoit Védie, Claudia de Toma (Biological resources center and tumor bank platform, AP-HP, Hôpital Européen Georges Pompidou); Sandrine Laboureau-Soares, Patrice Rodien, Frédéric Illouz, Katy Piton, Virginie Taillandier (Endocrinology), Odile Blanchet, Caroline Eymerit-Morin, Anne-Marie Croue, Marie-Christine Rousselet, Zidane Merzouka, Hélène Plouhinec, Stephanie Triau, Audrey Rousseau, Sophie Michalak (Biological resources center), Antoine Hamy, Stéphanie Mucci (Surgery, CHU Angers); Hervé Lefèbvre, Gaetan Prevost, Lucile Moreau-Grange, Adeline Savoure, Sandrine Menon, Karine Durand, Anne-Françoise Cailleux, Justine Bons, Hind Berrahmoune (Endocrinology), Jean-Christophe Sabourin (Pathology), Muriel Quillard (Biological research center, CHU Rouen); Emmanuel Sonnet (Endocrinology), Laurent Doucet (Pathology, CHU Brest); Françoise Borson-Chazot, Armelle Delahaye, Farida Smail, Lucie Parlant-Pinet (Endocrinology); Myriam Decaussin-Petrucci (Pathology), Laure Maillard, Jean-Christophe Lifante (Surgery, HCL, CHU Lyon Sud); Marie Batisse-Lignier (Endocrinology), Marc Berger (Biological resources center, CHU Clermont-Ferrand); Olivier Chabre (Endocrinology), Nathalie Sturm (Pathology), Pascal Mossuz, Philippe Lorimier, Gabriel Schummer (Biological resources center, CHU Grenoble); Philippe Chanson (Endocrinology), Catherine Guettier-Bouttier (Biological resources center, AP-HP, Hôpital de Bicêtre); Flora Fassio (Endocrinology), Valérie Rigau (Pathology, CHU Montpellier); Maëlle Le Bras, Anne-Laure Fournier, Ester Landau, Matthieu Pichelin, Violette Derry (Endocrinology), Cécile Girard, Estelle Verhille (Tumor bank platform), Karine Renaudin (Pathology), Eric Mirallié, Cécile Caillard, Christophe Ferron (Surgery, CHU Nantes); Guillaume Assié, Gaelle Lethielleux, Laurence Guignat, Rossella Libé, Lionel Groussin, Hélène Mosnier-Pudar, Seray Genc, Tanya Niati (Endocrinology), Béatrice Parfait, Benoit Girard, Michele Nigou, Dominique Pontoire (Biological resources center, APHP, Hôpital Cochin); Cécile Ghander (Endocrinology), Frédérique Tissier (Pathology, APHP, Hôpital de la Pitié-Salpétrière); Antoine Tabarin (Endocrinology), Philippe Merlio (Tumor platform bank, CHU Bordeaux); Bénédicte Decudier (Endocrinology), Danielle Diebold (Pathology, CHU Reims); Isabelle Guilhem (Endocrinology), Catherine Massart (Research clinic unit, CHU Rennes); Bertrand Goichot, Luz Siegel-Marc (Internal Medicine), Marie-Pierre Neu-Chenard (Pathology, CHU Strasbourg); Philippe Caron, Delphine Vezzosi, Céline Mouly (Endocrinology), Anne Brouchet (Pathology, CHU Toulouse); Peggy Renoult-Pierre, Lise Crinière, Anais Paillard, Maeva Dieu (Endocrinology), Serge Guyetant, Gaelle Fromont (Biological Resources Center), Caroline Gadras, Ludivine Diot, Sylvie Pineau, Marie Allet, Sarah Anceret (Biological resources center), Vincent Arnault, Sylvain Moriniere (Surgery, CHU Tours) ; Julien Hadoux, Ségolène Hescot, Jérôme Cartry (Endocrine Oncology), Abir Al-Ghuzlan (Pathology, Gustave Roussy Institute, Villejuif). They thank also all the members of the COMETE network, the Groupe d’Etude des Tumeurs Endocrines (GTE) and the TENgen network.
References
Footnotes
Contributors LBA, PP, LJCV and AB: performed the experiments. LBA, PP, JF, APGR and NB: designed the experiments. LBA, PP, LJCV, AB, JF, APGR and NB: collected the data and contributed to the interpretation of results. LA, JB, DD, IG, EB, CLL, CC, GC, CB, LF, CCB and APGR: provide and cared for studied patients. LBA, JF, APGR and NB: wrote the manuscript. APGR and NB: supervised the study. All authors approved the final version of the manuscript.
Funding This work has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 633983 and by the Institut National du Cancer and the Direction Générale de l’Offre de Soins (PRT-K 2014, COMETE-TACTIC, INCa-DGOS_8663). N. Burnichon received financial support from the Cancer Research for Personalized Medicine - CARPEM project (Site de Recherche Intégré sur le Cancer – SIRIC). The team is supported by the Ligue Nationale contre le Cancer (Equipe Labellisée). This work is part of the ‘Cartes d’Identité des Tumeurs (CIT) program’ funded and developed by the ‘Ligue Nationale contre le Cancer’ (http://cit.ligue-cancer.net).
Competing interests None declared.
Ethics approval Institutional Review Board (IRB 00003835, Comité de Protection des Personnes Ile de France IV).
Provenance and peer review Not commissioned; externally peer reviewed.