Article Text

Abstract
Background: We report here the genetic characterisation of a large five generation Chinese family with the phenotypic features of auditory neuropathy and progressive peripheral sensory neuropathy, and the genetic feature of X linked recessive inheritance. Disease onset was at adolescence (at an average age of 13 years for six affected subjects). The degree of hearing impairment varied from mild to severe, with decreased otoacoustic emissions; auditory brainstem responses were lacking from onset.
Methods: Two-point and multipoint model based linkage analysis using the MILNK and LINKMAP programs of the FASTLINK software package produced maximum two-point and multipoint LOD scores of 2.41 and 2.41, respectively.
Results: These findings define a novel X linked auditory neuropathy locus/region (AUNX1, Xq23–q27.3). This region is 42.09 cM long and contains a 28.07 Mb region with flanking markers DXS1220 and DXS8084, according to the Rutgers Combined Linkage-Physical Map, build 35. However, mutation screen of the candidate gene SLC6A14 within the region did not identify the causative genetic determinant for this large Chinese family.
- ABRs, auditory brainstem responses
- AMPA, alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate
- AN, auditory neuropathy
- DPOAEs, distortion product otoacoustic emissions
- MOS, medial olivocochlear systems
- NSRAN, non-syndromic recessive auditory neuropathy
- SLC, solute carrier family
- SNAP, sensory nerve action potential
- auditory neuropathy
- AUNX1
- Chinese pedigree
- gene mapping
- X linked recessive
Statistics from Altmetric.com
- ABRs, auditory brainstem responses
- AMPA, alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate
- AN, auditory neuropathy
- DPOAEs, distortion product otoacoustic emissions
- MOS, medial olivocochlear systems
- NSRAN, non-syndromic recessive auditory neuropathy
- SLC, solute carrier family
- SNAP, sensory nerve action potential
Auditory neuropathy (AN) is a hearing disorder characterised by a clinical syndrome including preservation of outer hair cell function with absent auditory brainstem responses (ABRs) and mild to profound hearing loss, accompanied by poor speech discrimination scores and poor understanding.1–4 Estimates of its prevalence varied from 0.5% (1/200) to 15% of patients with sensorineural hearing loss.5 Chinese people appear to have a high incidence of AN.4,6 So far several genes have been found to be associated with AN symptoms including the genes encoding peripheral myelin protein 22 (PMP22), myelin protein zero (MPZ), connexin32 (Cx-32), and early growth response 2 (EGR2).7 Three genes/loci were identified for non-syndromic AN: the OTOF gene (2p22–p23),8,9 the AUNA1 locus (13q14–21),10 and the mitochondrial 12SrRNA T1095C mutation.11 In previous clinical descriptions all three genes/loci contributed to the type II AN phenotype which involves the distal or peripheral auditory system (terminal dendrites, inner hair cells, and synapses). It was suggested that the OTOF gene was involved in a non-syndromic recessive auditory neuropathy (NSRAN) characterised by a set of special audiological features consistent with an autosomal recessive hearing disorder but without any other detectable peripheral neuropathy.8,9,12 The AUNA1 locus (auditory neuropathy, dominant, the first locus) maps to 13q14–21, which is linked to hearing impairment with preserved outer hair cell activity in the initial stage, a pattern of abnormality consistent with AN, and progressive hearing loss when outer hair cell activity from high and mid-frequency cochlear regions becomes impaired, consistent with combined partial sensory and neural hearing loss.3,10 A mitochondrial variant (12SrRNA T1095C) whose mutation causes AN symptoms without detectable peripheral neuropathy was identified recently in a Chinese patient.11 The T1095C mutation is predicted to disrupt an evolutionarily highly conserved A-to-U base pair at the P site of 12S rRNA with the I175V variant in the CO2 gene and the V112M variant in the ND6 gene, showing high evolutionary conservation, and may contribute to the phenotypic expression of the T1095C mutation in a Chinese AN patient. Identification of the underlying gene(s) for AN is one of the key challenges for understanding the molecular basis for different AN phenotypes.
Here we describe the detailed genetic characterisation of an extended five generation Chinese AN pedigree whose clinical features were previously reported.4 This Chinese AN pedigree provides strong evidence of X linked recessive inheritance. Using two-point and multipoint linkage analysis, the AUNX1 locus was localised between markers DXS1220 and DXS8084, which embrace a 42.09 cM region (28.07 Mb) according to the Rutgers Combined Linkage-Physical Map of The Human Genome (http://compgen.rutgers.edu/), build 35. However, mutation screening of the candidate gene within the region, SLC6A14 (AL034411, MIM 300444), did not identify the causative genetic determinant for the large Chinese AN family.
METHODS
Pedigree recruitment and phenotypic evaluation
The five generation Chinese family4 was ascertained from the Department of Otolaryngology, Head and Neck Surgery, at the Institute of Otolaryngology, PLA General Hospital, Beijing in November 2000. Informed consent, blood samples, and clinical evaluations were obtained from all participants in this family according to protocols approved by the Institutional Review Board. The diagnosis criteria for AN were defined as follows: onset of auditory complaints prior to or during adolescence, initially with hearing loss at low frequencies progressing to all frequencies, together with poor speech discrimination; normal or partially normal transient evoked otoacoustic emissions and distortion product otoacoustic emissions (DPOAEs); normal tympanometry; and abnormal ABRs and stapedial reflexes. In 2005, four individuals in the family were examined again to update their clinical records; audiological evaluation using the same methods as described previously4 and neurological investigation of cranial nerve function, motor activity, reflexes, and sensation were carried out.
Genotyping and linkage analysis
Genomic DNA was isolated from whole blood of 42 of 101 family members, who also underwent clinical auditory evaluation. Eighteen markers for the X chromosome (ABI Prism Linkage Mapping sets, version 2) were obtained from Perkin-Elmer Applied Biosystems (Foster City, CA). Twenty two pairs of locus specific primers were then purchased from Shenggong DNA Technologies (Shanghai, China) for fine mapping of the X linked region identified in the first stage linkage scan. Multiplex PCR was performed using PE9600 thermocyclers (Applied Biosystems), producing a final volume of 5 μl reaction mixture containing 30 ng of genome DNA, 1× PCR buffer, 0.2 mM of each dNTP, 3.0 mM MgCl2, 80 pmol of each forward and reverse primer, and 0.2 U of AmpliTaq Gold polymerase. Reactions were performed according to the manufacture’s instructions and the products were loaded onto a 6% denaturing polyacrylamide gel (7M urea) and visualised on an ABI 3700 sequencer. Alleles were analysed with ABI GeneMapper version 3.0. Two-point X chromosome linkage between the disease locus and the markers on the chromosome was evaluated using the MILNK program of the FASTLINK software package.13 The disease was hypothesised to be an X linked recessive disorder. Linkage analysis was carried out using a fully penetrant X linked model assuming a disease allele frequency of 0.0001. For the microsatellite marker loci, equal allele frequencies were used because a large sample of the ethnically matched control DNAs that could provide reliable estimates of the allelic frequencies for the reference population was lacking. It has been shown that when there are individuals within a pedigree who have not been genotyped, type I error can be increased14 when equal allele frequencies are used. In order to demonstrate that using equal marker allele frequencies did not increase type I error as almost every family member was genotyped within the pedigree, a sensitivity analysis was carried out by varying the marker allele frequency which segregates with the disease variant between 0.2 and 0.8. Multipoint X linkage analysis was carried out using the LINKMAP program of the FASTLINK package.15 Since FASTLINK uses the Elston-Stewart algorithm, it can handle large pedigrees but is limited in the number of loci. Therefore, a sliding window of two to five marker loci was used for the multipoint analysis. The number of markers used within the sliding window depends on the number of alleles at each marker locus which dictates how many marker loci can be analysed simultaneously using LINKMAP. For the multipoint analysis, genetic map distances were derived from the Rutgers Combined Linkage-Physical Map.16
Mutation screening
A candidate gene, SLC6A14 (AL034411), was selected for mutation screening. The SLC6A14 gene contains 14 exons and spans about 29 kb.17 Fourteen pairs of PCR primers for amplification of each exon of the SLC6A14 gene were designed using Primer3 software and synthesised based on intronic sequences provided by Shenggong DNA Technologies. All exons and/or exon-intron boundaries of the candidate gene were PCR amplified with PE9700 thermocyclers (Applied Biosystems). The PCR reactions were carried out in a total volume of 25 μl containing 2.5 mmol/l of each deoxynucleotide triphosphate (dNTPs; 3.0 μl each dNTP), 10 mmol of each primer, 1 U of AmpliTaq Gold polymerase (1 μl), 1.5 mmol/l MgCl2, 10× TE buffer (2.5 μl), and 100 ng of genomic DNA. PCR conditions were: 5 min pre-heating at 94°C, 30 s denaturation at 94°C, 30 s annealing at 58°C (slightly variable for each primer), 40 s extension at 72°C for 35 cycles, and then an additional 7 min extension at 72°C. After PCR amplification, 5 μl PCR products were separated on 1% agarose gel and purified using Millipore filter plates. Sequence analysis was performed for both affected and normal individuals of the family. DNA mutations were identified by aligning the sample sequences with GenBank databases using DNAStar software (DNASTAR, Madison, WI).
RESULTS
Phenotypic evaluation of the AN pedigree
The family, depicted as pedigree 1 in the previous report,4 originated from Shan Dong province in southeast China. Most of the family had been living in the same region for over 100 years. The auditory clinical data for family members with AN were described previously.4 The clinical records updated in 2005 show progressive hearing loss (fig 1), loss of outer hair cell function, and decrease in OAE amplitude in three re-examined affected individuals. The familial affiliations of these three subjects can be seen in fig 2, which shows the pedigree structure for the family members later subjected to haplotype analysis (described below). When tested in 2000, the proband (individual IV:24) had normal distortion-product otoacoustic emissions (DPOAEs) in the right ear, absent response at 1 and 2.5–6.3 kHz, and normal response over other frequencies. In 2005, the same subject had impairment in outer hair cell function at higher frequencies in the right ear, indicated by absent response at 3, 4, and 6 kHz, but normal DPOAE response in the range of 0.5–2 kHz. In the left ear, overall frequency responses were below the normal range of DPOAEs; the only positive responses were at 1, 2, and 4 kHz. Impaired outer hair cell function was also found in individuals IV:22 and IV:23. In addition, as shown in table 1, all three affected individuals who were re-examined, complained of extremity numbness lasting 2 months (IV:22), 6 months (IV:23), and 16 months (IV:24). All three subjects subsequently developed peripheral sensory neuropathy following the onset of the auditory disorder. The family clearly manifests the clinical features of type I AN phenotype which involves primary degeneration of the auditory nerve (that is, axons) accompanied by late onset peripheral neuropathy. Neurological examination revealed diffuse peripheral sensory neuropathy. Nerve conduction evaluation showed that the sensory nerve action potential (SNAP) is not induced in the bilateral median, ulnar, peroneal, tibial, and sural nerves in IV:22, in the left median and ulnar nerves in IV:23, and in the right median and bilateral sural nerves in IV:24. Sensory nerve conduction velocity is slightly decreased (31.9∼48.5 m/s) in the right median (48.5 m/s), ulnar (39.4 m/s), and bilateral sural nerves (L: 31.9 m/s; R: 34.1 m/s) in IV:23, and in the left median (47.1 m/s) and bilateral ulnar (L: 44.8 m/s; R: 38.2 m/s) nerves in IV:24. Furthermore, the SNAP amplitudes in the above nerves are notably decreased, being 0.4 μV in the right median, 1.0 μV in the right ulnar, and 0.8 and 0.6 μV in the left and the right sural nerves in IV:23, and 3.7 μV in the left median and 3.1 and 0.7 μV in the left and the right ulnar nerves in IV:24. Nevertheless, motor nerve conduction is normal for all three affected individuals. These results indicate diffuse axonal sensory neuropathy.
Updated clinical data for the three affected members
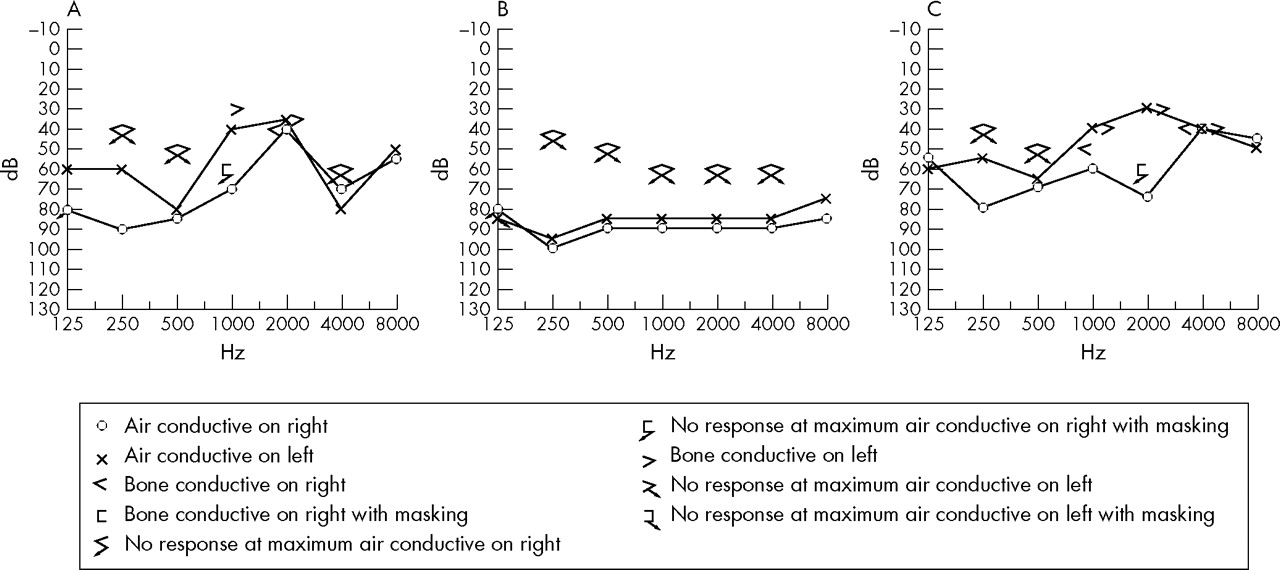
Updated audiograms for the three affected members (A: IV:22, B: IV23, and C: IV:24).
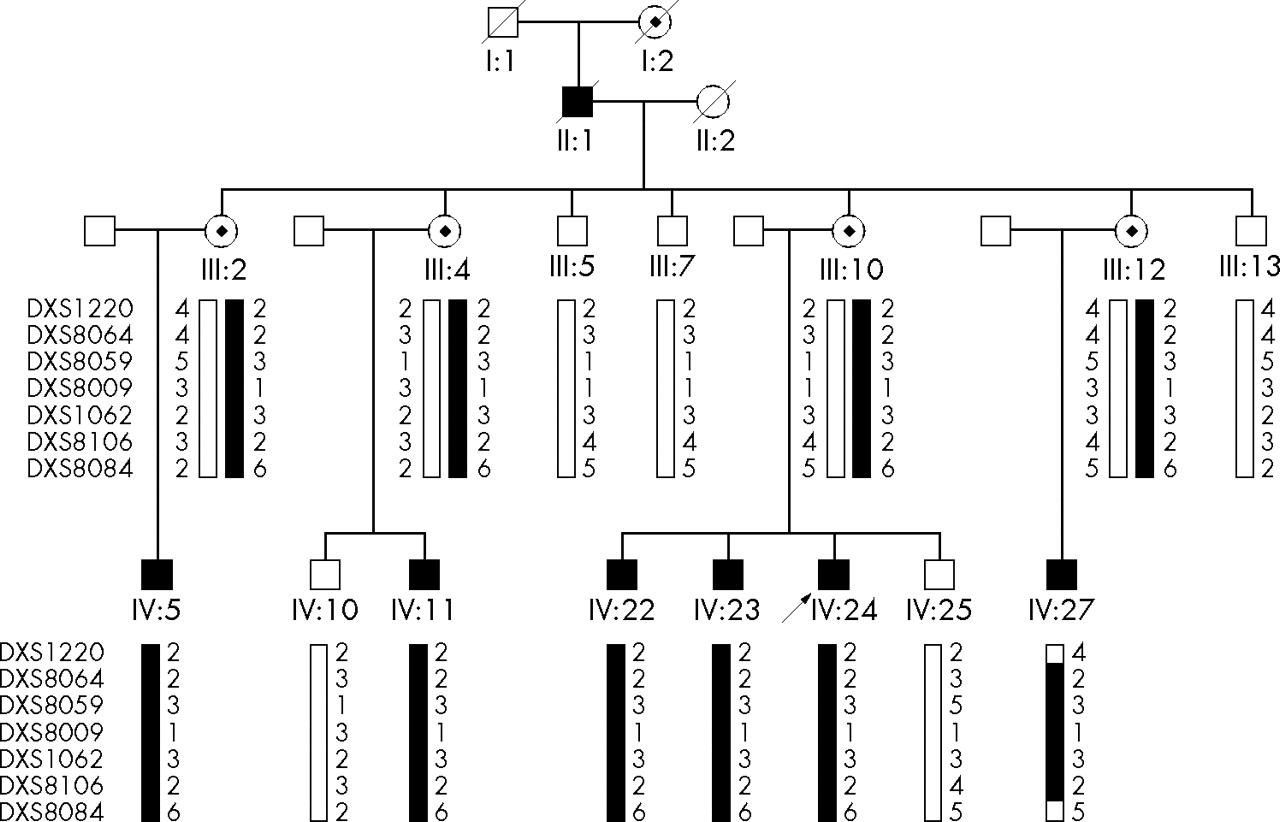
Haplotype analysis in an AUNX1 kindred with auditory neuropathy. Genotyping results for markers DXS1220, DXS8064, DXS8059, DXS8009, DXS1062, DXS8106, and DXS8084 are shown. Haplotypes were constructed on the basis of the minimum number of recombination events between the markers. The disease haplotype shared by all affected individuals is indicated by the filled vertical bar and the normal haplotype is denoted by an open vertical bar. Recombination events were observed in subject IV:27 and defined the critical AUNX1 gene location between markers DXS1220 (centrometric) and DXS8084 (telemetric) in Xq23–27.3.
Model based linkage analysis
The results of two-point linkage analyses on the X chromosome (Xq23–27.3) with selected markers are shown in table 2. The maximal two point LOD score was 2.41 at a recombination rate (θ) of 0 for DXS1001, DXS1212, and DXS1211. A sensitivity analysis was carried out to see if using equal allele frequencies in the analysis would bias the results. When the allele frequency for the allele segregating with the disease phenotype was varied between 0.2 and 0.8, the maximum LOD score remained at 2.41 at θ = 0 for markers DXS1001, DXS1212, and DXS1211. In addition, the use of different assumed marker allele frequencies did effect any of the resulting LOD scores. Therefore, using equal allele frequencies for the marker loci did not increase the LOD scores for the particular sample (table 2).
Two-point LOD scores between the X linked AN locus and chromosome markers
For multipoint linkage analysis, markers DXS1220 and DXS8084 reached negative infinity. For the marker flanking DXS1220, marker DXS8064, the LOD score reached its maximum of 2.41 and remained unchanged for the following markers: DXS8067, DXS1001, DXS8059, DXS1212, DXS8057, DXS8093, DXS8009, DXS1047, DXS1062, DXS1211, DXS8013, DXS984, DXS1205, DXS1227, and DXS8106.
Haplotype transmission pattern analysis further validated the mapping of the novel AN locus, AUNX1. Figure 2 shows the haplotypes of seven markers (DXS1220, DXS1001, DXS8059, DXS8009, DXS1062, DXS8106, and DXS8084). According to the region shared by the affected male (IV:5, IV:11, IV:22, IV:23, and IV:24) and female carriers (III:2, III:4, III:10, and III:12), we situated the critical AUNX1 gene within a region spanned by markers DXS1212 and DXS8084. Two recombination events were found in subject IV:27, which defined a region of 42.09 cM flanked by markers DXS1220 (centromeric) and DXS8084 (telomeric) in Xq23–27.3 (fig 3). The locus was nominated as AUNX1 (auditory neuropathy X linked recessive locus 1) and the family is designated as the AUNX1 family (http://www.gene.ucl.ac.uk/nomenclature/). AUNX1 is the first locus identified for X linked recessive AN.

{kind=link}
{kind=link}
{kind=link}
Ideogram of chromosome X with Geimsa banding and localisation of the Xq23–q27.3 AN region. The genetic map within the region is shown, and the likely location of the putative AUNX1 gene is indicated by a vertical bar (from DXS1220 to DXS8084).
DISCUSSION
In the present study, we performed detailed genetic characterisation of a large five generation Chinese family with early onset AN and later onset of peripheral sensory neuropathy without detectable motor nerve conduction abnormality, leading to definition of the first X linked locus (Xq23–27.3) for the type I AN phenotype. This hearing loss locus, AUNX1, is a novel linkage relating to a distinct phenotype of the sex linked hearing disorders identified so far.
In the past decade, great progress has been made in identifying genes responsible for non-syndromic hearing impairment. To date, more than 100 loci for non-syndromic hearing loss have been mapped to the human genome. Furthermore, at least 40 genes have been cloned and an equivalent number mapped for dominant, recessive, or X linked hearing loss (Hereditary Hearing Loss Homepage, http://webhost.ua.ac.be/hhh/). Genes contributing to hereditary hearing loss encode a wide variety of protein classes: from myosins and other cytoskeletal proteins, channel and gap junction components, to transcription factors, extracellular matrix proteins, and even some with unknown functions. Almost all non-syndromic hearing loss genes express in the inner ear, especially in basilar membrane, bony spiral lamina, inner hair cells, outer hair cells, Reissners’ membrane, spiral ligament, stria vascularis, and tectorial membrane (Hereditary Hearing Loss Homepage, http://webhost.ua.ac.be/hhh/). The genes that express differentially in the auditory nerve and/or the synapses between inner hair cells and the auditory nerve as well as in the pathway of medial olivocochlear systems (MOS) remain to be fully explored.
To summarise, few genes/loci have been found except for the OTOF gene, the AUNA1 locus, and a mitochondrial variant (12SrRNA T1095C), for understanding the molecular basis for different types of AN. Mutations in the otoferlin (OTOF, MIM 603681) gene were suggested as the causative gene for NSRAN, implying that the OTOF gene may play a synaptotagmin-like role in IHC synaptic vesicle fusion.18 Mutation in the mitochondrial variant T1095C in the 12SrRNA gene may be associated with AN in a Chinese AN patient. Two novel variants, the I175V mutation in the CO2 gene and the V112M mutation in the ND6 gene, may play a role in the pathogenesis of this disorder. Furthermore, it seems that the otoferlin gene may not be involved in the phenotypic expression of the T1095C mutation in this Chinese AN patient.11
The lesions associated with the AUNX1 locus in our Chinese family can be reasonably hypothesised to occur where auditory nerve demyelination and/or synapse dysfunction between inner hair cells and the auditory nerve takes place, or in the MOS pathway19 with normal outer hair cell function. The candidate gene, SLC6A14, within the AUNX1 locus, might be a good candidate because its function may relate to the aetiology of AN. Because the AN patients had intact outer hair cell function but absent synchronous neural activity (action potentials) on acoustically evoked auditory brainstem response, possible lesion sites in AN include inner hair cells, the synapse between hair cells and the auditory nerve, neural dendrite or axon, myelin sheath, or spiral ganglion cells; disruption of neurotransmitter transportation may occur in some of these sites. It is interesting that the SLC6A14 gene is a member of a solute carrier family (SLC), a type of neurotransmitter transporters, and is also a major gene in hereditary recessive hearing impairment.20–22 In previous studies, the SLC26A4 gene, which encodes an anion transporter, was proved to be the causative factor in Pendred syndrome (MIM 274600), DFNB4 (MIM 600791), and enlarged vestibular aqueduct syndrome (MIM 603545).23,24 In our study, we first selected SLC6A14, one of the family members of SLC, as a candidate gene of the AUNX1 locus. However, exon screening of the SLC6A14 gene did not identify any mutation in the Chinese AN family. Another promising candidate gene that maps to the AUNX1 locus is GRIA3 (MIM 305915), one of the glutamate receptor genes.25,26 Glutamate receptors are the predominant excitatory neurotransmitter receptors in the mammalian brain and are activated in a variety of normal neurophysiologic processes.27,28 These receptors are heteromeric protein complexes with multiple subunits, each possessing transmembrane regions, and all arranged to form a ligand gated ion channel. This gene belongs to a family of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors. SOX3 (MIM 313430) within the genetic region is also a good candidate gene and encodes a member of the SOX (SRY related HMG box) family of transcription factors involved in the regulation of embryonic development and in the determination of cell fate. The encoded protein may act as a transcriptional regulator after forming a protein complex with other proteins. Mutations in this gene have been associated with X linked mental retardation with growth hormone deficiency.
In conclusion, we have reported a novel locus for AN with X linked recessive inheritance and with onset during adolescence, low frequency hearing loss progressing to all frequencies, poor speech discrimination, normal or nearly normal TEOAEs and DPOAEs in the initial stage, normal tympanometry and absent ABRs and stapedial reflex in all affected males with late onset of periphery neuropathy. Further positional cloning of the X chromosome AN gene(s) is required through investigating more X linked families, shortening the region of the AUNX1 locus, and screening the candidate genes, which will contribute to better understanding of the molecular mechanism(s) of the AN phenotype.
ACKNOWLEDGEMENTS
We thank four reviewers for their helpful comments on an earlier version of the manuscript. We also thank the patients and their families for their cooperation during this work, and the China Association of the Handicapped for their assistance in our investigation.
REFERENCES
Footnotes
-
↵The first three authors (Q J Wang, Q Z Li, S Q Rao) contributed equally to this work
-
This work was supported by grants from the National High Tech Development Project (grant nos. 2004AA221080 and 2003AA205070), the National Natural Science Foundation of China (grant nos. 30370782, 30370798, 30470956, and 30570424), the Beijing Science Technology Project (grant no. H020220020610), the Foundation of National Excellent Doctoral Thesis (grant no. 2004No63), the Heilongjiang Province Department of Education Outstanding Overseas Scientist grant (grant no. 1055HG009), and the US National Institutes of Health National Institute of Deafness and Other Communication Disorders (grant no. R01-DC03594)
-
Conflicts of interests: none declared