Article Text

Abstract
Background: Subtelomere fluorescence in situ hybridisation (FISH) analysis has increasingly been used as an adjunct to routine cytogenetic testing in order to detect small rearrangements. Previous reports have estimated an overall abnormality rate of 6%, with a range of 2–29% because of different inclusion criteria.
Methods: This study presents data compiled from 11 688 cases referred for subtelomere FISH testing in three clinical cytogenetic laboratories.
Results: In this study population, the detection rate for clinically significant subtelomere abnormalities was approximately 2.5%, with an additional 0.5% detection of presumed familial variants. Approximately half of the clinically significant abnormalities identified were terminal deletions, the majority of which were de novo. Most of the remaining cases were unbalanced translocations between two chromosomes or two arms of the same chromosome. Approximately 60% of the unbalanced translocations were inherited from a parent carrying a balanced form of the rearrangement. Other abnormalities identified included tandem duplications, apparently balanced translocations, partial deletions, and insertions. Interestingly, 9 cases (0.08%) were found to have interstitial deletions of non-telomeric control loci, either BCR on 22q or PML on 15q. The most common clinically significant imbalances found were deletions of 1p, 22q, 4p, 9q, 8p, 2q and 20p. The most common familial variants were a deletion or duplication of 10q, deletion of 4q, deletion of Yq, and duplication of X/Yp onto Xq.
Conclusions: This study of subtelomere rearrangements is a 20 fold increase in number over the previously reported largest study and represents an unbiased analysis of subtelomere rearrangements in a large, unselected patient population.
- CNP, copy number polymorphism
- FISH, fluorescence in situ hybridisation
- subtelomere
- telomere
- fluorescence in situ hybridisation
- variant
- mental retardation
Statistics from Altmetric.com
Over the past several years, the use of genome wide subtelomere screening has increasingly been used as an adjunct to routine cytogenetic testing and has already been incorporated into recommendations for the evaluation of individuals with unexplained mental retardation/developmental delay.1,2 To assess the usefulness of this testing, Biesecker3 reviewed 14 previously reported studies (comprising 1718 subjects) that performed subtelomere analysis. These studies showed an overall abnormality rate of 6%, with a range of 2–29% in individual studies. The large variance in frequency of abnormalities observed is most likely due to the different criteria used for inclusion and the sample size in each of the studies. In the largest single study of subtelomere abnormalities to date in patients with a normal karyotype, Knight et al4 examined 466 individuals and reported a frequency of 7.4% (95% confidence interval (CI) 4.4 to 10.4%) in individuals with moderate to severe mental retardation and 0.5% (95% CI 0 to 1.6%) in individuals with mild mental retardation.
Interpreting the results from subtelomere testing can be complicated by the fact that in addition to those subtelomere rearrangements that are the most likely cause of the phenotype, there are also deletions or duplications of the subtelomere regions that appear to be benign familial variants, where an affected proband has an imbalance that is subsequently identified in one of the phenotypically normal parents. Many examples of such cases have previously been reported in the literature, including deletions of 2q, 4q, 10q, 14q, and 17p, and duplications of 4q, 6p, and 10q.5,6,7,8,9,10 These findings underline the importance of follow up parental analysis when a subtelomere abnormality is identified in an affected proband to determine the clinical significance of the results.
In this study, subtelomere screening was carried out on 11 688 individuals who were referred to a diagnostic cytogenetics laboratory for fluorescence in situ hybridisation (FISH) testing as part of a clinical evaluation. Any unbalanced subtelomere rearrangements that were identified were categorised as causative of the phenotype or benign familial variants whenever parental analysis was performed and this interpretation could be made. This study of subtelomere rearrangements is a 20 fold increase in number over the previously reported largest study, and represents an analysis of the frequency and pattern of subtelomere rearrangements, both pathogenic and variants, in a large, unselected patient population.
METHODS
Cases included in this retrospective study were received by the clinical cytogenetics laboratory at either Genzyme Genetics (Sante Fe, NM, USA), Laboratory Corporation of America (Research Triangle Park, NC, USA) or the University of Chicago (Chicago, IL, USA) for subtelomere FISH screening. Institutional review board procedures were followed by each institution. Samples were received from a variety of healthcare providers, including but not limited to geneticists, paediatricians, and paediatric neurologists. The majority of subjects were referred due to developmental delay or mental retardation with or without dysmorphic features; however, a wide range of indications was noted, such as behavioural disorder, autism, or growth delay. Chromosome analysis was recommended before subtelomere FISH studies were performed. Banding resolution of studies performed in these three laboratories was routinely 550 bands and above. Cases with cytogenetically visible abnormalities were excluded from this study. The indication for referral for cases in which a subtelomere abnormality was identified is listed in table 1.
Subtelomere rearrangements listed by abnormality type
Peripheral blood samples were cultured and harvested according to standard protocols. Genome wide subtelomere FISH analyses were carried out using either the ToTelVysion assay (Vysis/Abbott, Inc., Downers Grove, IL, USA) or the Chromoprobe Multiprobe-T System (Cytocell Technologies, Ltd., Cambridge, UK), both of which consist of 41 telomere probes.11 The Cytocell Chromoprobe system was used for only a few specimens in one laboratory at the beginning of the study. Thereafter, all analyses were carried out using the Vysis ToTelVysion probe panel. The majority of the probes in the two systems target the same loci; additional information can be found on the manufacturers’ websites (www.vysis.com and www.cytocell.com). The manufacturers’ instructions were followed and 5–10 metaphase cells were analysed for each hybridisation area.
Hybridisation signals were evaluated for hybridisation to the correct subtelomere region as well as signal intensity, judged by direct microscopic visualisation. If the signal intensity was consistently unequal between homologues, the signal was defined as diminished or duplicated, depending on the pattern observed in interphase cells. A diminished signal represents a partial deletion where the breakpoint is contained within the clone and only involves a portion of the subtelomere probe target sequences. Hybridisation signals observed on non-target chromosomes were characterised as cross hybridisation if the signal was observed on a pair of homologues with similar intensity that was reduced relative to the signal on the target chromosome.
All abnormalities identified were confirmed with either repeat FISH analysis using a single subtelomere probe or targeted G banded chromosome confirmation. Parental FISH analyses of probands with subtelomere rearrangements were performed when possible to determine whether the abnormality was inherited or de novo. For parental analysis, targeted FISH was performed, using only the probes that showed abnormal hybridisation patterns in the proband. Rearrangements were characterised as familial variants when an unbalanced subtelomere rearrangement was inherited from a parent with the same imbalance and a normal phenotype, as reported by the referring physician. When parental analysis was not available, a rearrangement was designated as a possible variant if the same imbalance was observed and characterised as a familial variant in at least one other case.
RESULTS
Genome wide subtelomere FISH analysis was used in this study to analyse 11 688 peripheral blood specimens. As listed in table 1, 357 abnormalities were identified in 355 individual probands, an overall abnormality rate of 3.0%. Two probands were found to each have two presumably unrelated subtelomere rearrangements, one of which was clinically significant and the other a familial variant. The male:female sex ratio was virtually even at 169:170. The average age at diagnosis was 7 years (range 1 day to 70 years).
The most common subtelomere rearrangements observed were deletions and unbalanced translocations. As summarised in table 2, terminal deletions were identified in 175 cases (49.3% of all rearrangements found); an additional 19 cases (5.4%) showed a partial deletion. Also shown in table 2, 145 cases (40.8%) had unbalanced derivative chromosomes that were divided into three categories: (a) a derivative chromosome with both a duplication and a deletion of subtelomere regions (108 cases, 30.4%); (b) a derivative chromosome with only a duplication of subtelomere sequences translocated distal to an intact subtelomere region of another chromosome (20 cases, 5.6%); or (c) a derivative chromosome with a duplication onto the short arm of an acrocentric chromosome (17 cases, 4.8%). Representative images from each of these types of subtelomeric rearrangements are shown (fig 1). Other subtelomeric rearrangements were observed at lower frequencies; four (1.1%) tandem duplications, four (1.1%) apparently balanced reciprocal translocations, and one (0.3%) apparently balanced insertion were found.
Summary of each type of subtelomere abnormality identified in this study and the inheritance patterns observed when parental studies were performed
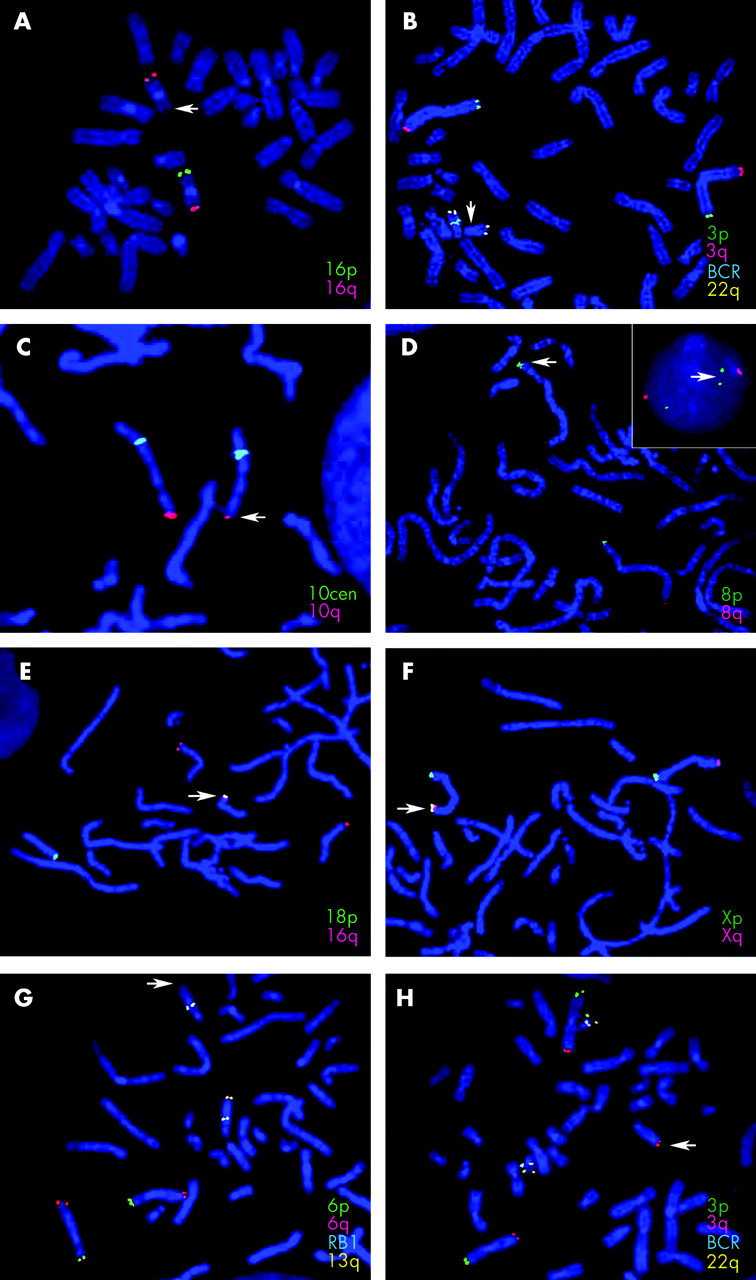
Representative FISH images of the various subtelomere rearrangements identified in this study. Arrows mark the abnormal chromosome. The probes used are listed for each image in the colour that corresponds to the colour of their hybridisation signal. (A) 16p subtelomere deletion; (B) deletion of the BCR control probe on chromosome 22q; (C) partial deletion of the 10q subtelomere; (D) duplication of the subtelomeric region of 8p shown on metaphase chromosomes and in an interphase nucleus (inset); (E) translocation of the 16q subtelomere probe to the short arm of chromosome 18, distal to the 18p subtelomere probe; (F) additional signal for Xp/Yp located on Xq adjacent to the X/Yq subtelomere probe signal; (G, H) unbalanced translocation between the subtelomeric regions of 3q and 13q resulting in monosomy for 13q (G) and trisomy for 3q (H).
Interestingly, nine interstitial deletions of non-telomeric regions were identified in this study by noting the absence of a signal for one of the control probes used in the subtelomere probe panel. Eight interstitial deletions involved the 22q11.2 BCR probe, which is used as a control probe in the mixture containing the 22q subtelomere probe. A FISH probe for the DiGeorge/velocardiofacial syndrome region at 22q11.2 was subsequently tested and was normal in all eight cases. In addition, one case showed an interstitial deletion of sequences targeted by the 15q24 PML probe used as a control probe in the mixture containing the 15q subtelomere probe. These cases may represent new microdeletion syndromes and comparisons of the phenotype of these individuals and further characterisation of their deletions is in progress.
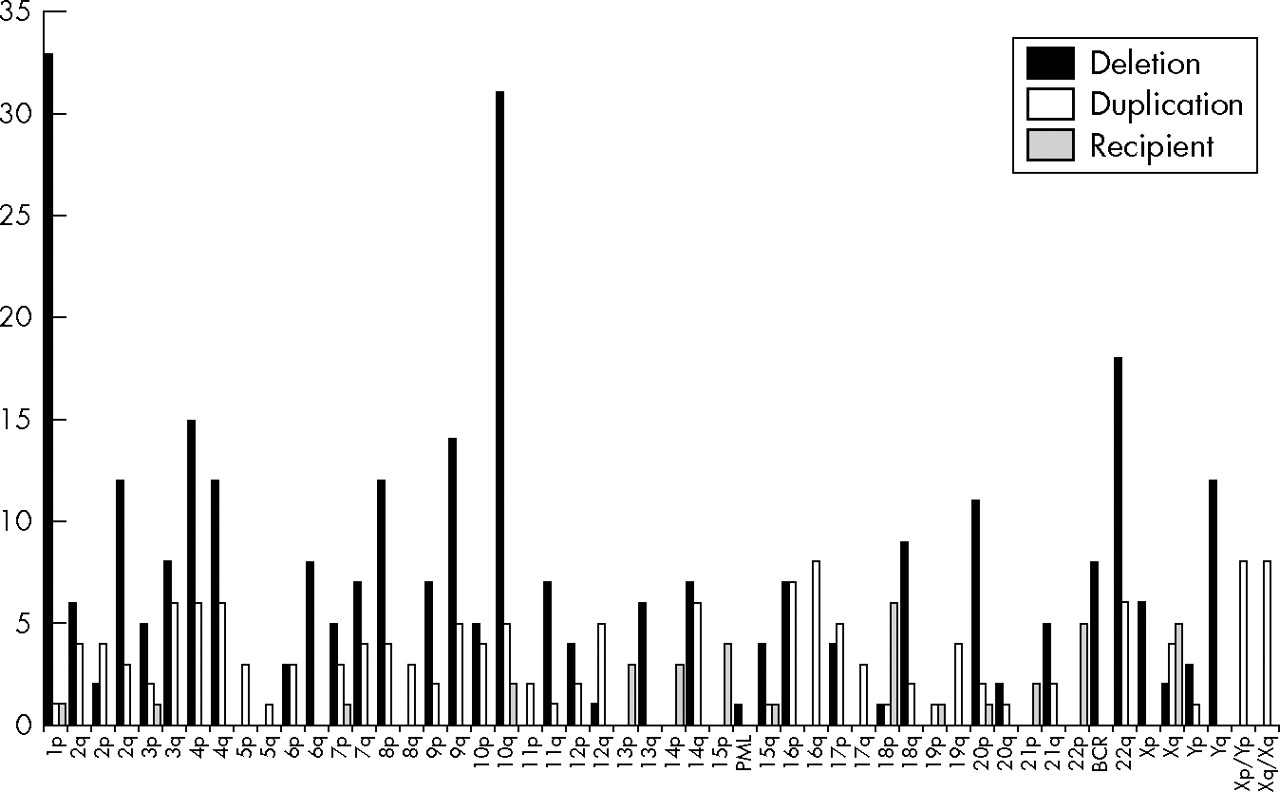
The overall frequency of rearrangements found for each subtelomere region is shown in fig 2, classified by whether the rearrangement led to a duplication or deletion of the chromosome end, or whether the end remained intact but had additional material from another chromosome end added distally.

{kind=link}
{kind=link}
Number of subtelomeric rearrangements found involving each chromosome region. Solid bars, rearrangements in which the subtelomere region was monosomic; white bars, rearrangements in which the subtelomere region was trisomic; grey bars, rearrangements in which the subtelomere region targeted by the probe was intact but an additional subtelomere region was present distally. PML and BCR deletions represent interstitial deletions detected by the absence of the control probe signal for chromosomes 15 and 22, respectively.
Parental FISH studies were recommended for all patients in whom a subtelomere rearrangement was found. Targeted studies using the probes showing an abnormality in the proband were performed on 136 sets of parents. Table 2 summarises the inheritance pattern observed for each type of abnormality. The majority of the terminal deletions (48 of 60; 80%) were found to be de novo. Twelve (20%) were inherited from a parent who carried the same deletion; 10 of these parents were reported by the referring physician to be phenotypically normal and two were reported to have abnormalities suggestive of a genetic imbalance. Parental follow up studies were available for 10 partial deletions, of which one (10%) was found to be de novo, with the remaining nine (90%) inherited from a parent with the same partial deletion. All of these parents were reported to be phenotypically normal. Only two of the interstitial deletions had complete parental follow up studies; both (100%) were found to be de novo.
Of the four cases with tandem duplications, parental studies were available for one; this dup(8)(pter) was found to be inherited from a phenotypically normal mother with the same duplication.
Unbalanced translocations involving more than one subtelomere region may result from meiotic segregation of a balanced rearrangement carried by a parent. Parental studies showed that of the 63 rearrangements involving more than one chromosome end, 29 (46%) were inherited from a parent carrying a balanced form of the rearrangement. Of the different types of rearrangements included in this group, inheritance from a balanced parent was less frequent for the derivatives containing a duplication only (two of 12; 17%) or a duplication onto the short arm of an acrocentric chromosome (one of 9; 11%) than for the derivatives containing both a duplication and a deletion (26 of 42; 62%).
The same unbalanced translocation observed in a proband was found in a parent in nine of 12 (75%) cases containing a duplication only and in two of 9 (22%) cases with a duplication onto the short arm of an acrocentric chromosome; all of these carrier parents were reported to be phenotypically normal. One case with an abnormal X chromosome containing a deletion of Xp and a duplication of Xq/Yq was found to be inherited from a mother with the same rearrangement; however, the mother was reported to have an abnormal phenotype.
Of 63 unbalanced translocations, 22 (35%) were found to be de novo. This includes 15 of 42 cases (36%) with both a duplication and deletion, 1 of 12 cases (8%) with a duplication only, and 6 of 9 cases (67%) with a duplication onto the short arm of an acrocentric chromosome. No parental follow up studies were available for the probands with apparently balanced insertions or translocations.
The discovery of so many unbalanced subtelomere rearrangements found in reportedly normal parents complicates the interpretation of the clinical significance of individual abnormal subtelomere findings. In an attempt to clarify which subtelomere regions may have imbalances that do not result in phenotypic abnormalities, the rearrangements found in this study were classified as probably clinically significant or not. A rearrangement was considered a familial variant if parental FISH studies were performed and a phenotypically normal parent was shown to carry the same unbalanced rearrangement as the affected proband. When parental analysis was not available, a rearrangement was classified as a possible variant if the same rearrangement was shown to be a familial variant in at least one other unrelated case. All other rearrangements were assumed to be clinically significant. Of 357 abnormalities found, 31 were considered familial variants and 25 possible variants, a total of 56 likely variants; 16% of the total number of abnormalities found. The remaining 84% (301) of the abnormalities found were assumed to be clinically significant. However, without performing family studies or fine mapping to determine the actual size of the deleted or duplicated regions for the 24 cases categorised as possible variants, it is not accurately known if these rearrangements are benign variants versus clinically significant. Therefore, the number of clinically significant abnormalities identified could be under-represented.
Table 3 shows the rearrangements classified as likely variants in this study. The most common variants observed involved rearrangements of the 10q subtelomere region. Fourteen cases had a partial deletion for the 10q subtelomere probe region, one case had a deletion of the entire 10q subtelomere probe region, and two had apparent tandem duplications of the 10q probe region.
Subtelomeric rearrangements classified as variants
Three unbalanced translocations were found to be familial variants in more than one family. Five cases were found with an extra Xp/Yp subtelomere signal on the long arm of the X chromosome adjacent to the Xq/Yq subtelomere probe signal (both maternal and paternal inheritance were observed), three cases had an additional copy of the 4p subtelomere probe region on the short arm of chromosome 22, and five cases had an extra hybridisation signal for the 16q subtelomere region present on 18p adjacent to the 18p subtelomere probe signal. Other variants observed in more than one family included deletions of 4q, 21q, and Yq, and partial deletions of 14q.
Of particular interest are the two patients who each had two unrelated subtelomere abnormalities. One proband was found to have a deletion of 16p as well as a partial deletion of 10q (122 and 180 in table 1). Parental studies showed that the 16p deletion was de novo, but the partial 10q deletion was inherited from the mother, who showed the same diminished 10q signal. The second proband was found to have a deletion of 4q and a derivative 8 from a translocation between 4p and 8p (68 and 292 in table 1). Parental studies were not available for this family, but subtelomere FISH analysis of a sibling showed the same derivative 8 as the proband, but no deletion of 4q. The two siblings were reported to have similar phenotypes.
DISCUSSION
This study of 11 688 individuals demonstrates that rearrangements of the subtelomere regions contribute significantly to idiopathic mental retardation. The individuals tested here were referred for clinical subtelomere testing by a broad spectrum of medical specialties, including paediatrics, neurology, and genetics. The clinically significant abnormality rate of 2.6% detected in this cohort is similar to that previously reported for routine cytogenetic and fragile X testing of probands with unexplained developmental disabilities.4,12 Thus, subtelomere testing is a vital diagnostic tool for individuals with unexplained mental retardation or developmental delay. In addition, as 46% of the clinically significant subtelomere alterations identified in this study were inherited from a parent carrying the balanced form of the rearrangement, these findings have obvious implications for recurrence risk estimates and genetic counselling.
Previous studies examining the incidence of subtelomeric rearrangements have reported a frequency of 2–29%.3,13 The largest study published to date, on individuals with normal routine chromosome analysis, showed a detection rate of 7.4% in individuals with moderate to severe mental retardation and 0.5% in individuals with mild mental retardation.4 In the present study, the inclusion of all patients referred to a clinical cytogenetics laboratory for subtelomere testing, regardless of the severity of mental retardation or presence of dysmorphic features, may explain the lower frequency of abnormalities. Additionally, as subtelomere imbalances are identified in individuals with only developmental delay, mild mental retardation, and/or mild dysmorphic features, the population of individuals being tested could also be expanding, thus leading to an overall lower frequency. However, by using samples that were analysed as part of routine clinical care and not specifically solicited according to strict clinical criteria, this study better represents the incidence in the general delayed patient population.
A wide range of abnormalities, including deletions, duplications, and unbalanced translocations were observed in this study. One of the most important categories is the unbalanced translocations that involve a subtelomere region and the short arm of an acrocentric chromosome. These cases demonstrate that apparent acrocentric short arm polymorphisms by G banding analysis can actually represent clinically significant genomic imbalances, underlining the importance of using subtelomere FISH for individuals with an abnormal clinical phenotype and a normal karyotype.
Deletions were observed much more frequently than duplications in this study, as was found in previous reports.4,14 Duplications are difficult to identify using metaphase FISH analysis. Therefore, the incidence of subtelomeric duplications may be higher, but is under-represented by current testing methods. As more studies are performed using newer technologies, such as array based comparative genomic hybridisation or quantitative DNA based assays, which are better suited to detect duplications, the true frequency of these abnormalities will be revealed.
In this study, 1p and 22q subtelomere deletions were the most frequent clinically significant imbalances observed. Both of these chromosomal regions are difficult to analyse using routine G banding analysis, which could contribute to this finding. The increased frequency of these deletions suggests there may be an underlying genomic mechanism contributing to their recurrence. To date, no common mechanism that accounts for these recurring deletions has been identified.
One of the difficulties in interpreting results from subtelomere testing is determining the clinical significance of the findings. Once an abnormality is identified, additional familial studies (preferably performed in the same laboratory that first identified the imbalance) are critical for accurately interpreting the results for the proband and for estimating recurrence risks for other family members. In this study, parental analysis was not available for many of the abnormalities identified, in spite of strong recommendations by the laboratories. Therefore, some of the deletions or derivative chromosomes with additional material from another subtelomeric region that were categorised as clinically significant may actually be familial variants.
Partial deletions of the 10q subtelomere region were commonly observed in this study. Owing to the high frequency of this observation, a partial deletion of the 10q subtelomere clone is most likely a common polymorphism, much like the common polymorphism previously described for the 2q telomere region.8 However, parental analysis is still recommended for these cases until more accurate genotype/phenotype correlations can be defined. As partial deletions or duplications of the 10q subtelomere clone were observed frequently in this study and have also been encountered in other studies,7,9,15 one commercially available assay used in this study (ToTelVysion) was modified to avoid detection of this polymorphism.
The variants that have been reported in studies using subtelomere probes are interesting. Historically, most cytogenetic rearrangements that are identified in an affected proband and subsequently in an unaffected parent are deemed probably unrelated to the clinical findings in the proband. The same holds true for our current understanding of subtelomere rearrangements. In addition, with the advent of new technologies to examine copy number changes across the entire genome, several reports have now documented the existence of genome wide copy number polymorphisms (CNPs).16–18 A comparison of the subtelomere clones with variants identified in this study against a database of previously reported CNPs (http://projects.tcag.ca/variation/) revealed that deletions of 4q,9,17 10q,9,15 and 14q,17 and duplications of 10q9,15 have been previously observed. However, the possibility still exists that the difference in phenotypic expression between the parent and the affected child could be due to subtle differences in the rearrangement, modifier genes, genes present on the normal homologue, epigenetic effects or other, not yet described phenomena.
In order to gain accurate knowledge about the consequences of specific subtelomere rearrangements, additional mapping studies for each subtelomere region are necessary to define the size of a deletion or duplication that has a phenotypic effect compared with those that are tolerated without clinical effects. This mapping information, together with the phenotypic observations, will allow the development of genotype/phenotype correlations to aid in the diagnosis, prognosis, and clinical management of individuals with subtelomere rearrangements. In the near future, through the use of DNA based methods for detecting genomic imbalances, such as array CGH, in conjunction with expanded clone coverage for the subtelomere regions, these determinations will be available as a more efficient and comprehensive diagnostic test.
Acknowledgments
The authors would like to thank the members of the clinical cytogenetics and FISH laboratories at Genzyme Genetics, Laboratory Corporation of America and the University of Chicago for excellent technical support.
REFERENCES
Footnotes
-
Published Online First 30 September 2005
-
Competing interests: J B Ravnan and J H Tepperberg have previously been compensated for speaking about subtelomere rearrangements at a symposium sponsored by Vysis/Abbott, Inc. C L Martin has an educational research grant from Vysis/Abbott, Inc. for the development of a website for genotype/phenotype correlations of subtelomere rearrangements.
-
The first two authors contributed equally to this work.