Article Text

Abstract
Autosomal dominant optic atrophy (ADOA) is genetically heterogeneous, with OPA1 on 3q28 being the most prevalently mutated gene. Additional loci are OPA3, OPA4, and OPA5, located at 19q13.2, 18q12.2, and 22q12.1–q13.1, respectively. Mutations in the WFS1 gene, at 4p16.3, are associated with either optic atrophy (OA) as part of the autosomal recessive Wolfram syndrome or with autosomal dominant progressive low frequency sensorineural hearing loss (LFSNHL) without any ophthalmological abnormalities. Linkage and sequence mutation analyses of the ADOA candidate genes OPA1, OPA3, OPA4, and OPA5, including the genes WFS1, GJB2, and GJB6 associated with recessive inherited OA or dominant LFSNHL, were performed. We identified one novel WFS1 missense mutation E864K, c.2590G→A in exon 8 that co-segregates with ADOA combined with hearing impairment and impaired glucose regulation. This is the first example of autosomal dominant optic atrophy and hearing loss associated with a WFS1 mutation, supporting the notion that mutations in WFS1 as well as in OPA1 may lead to ADOA combined with impaired hearing.
- ADOA, autosomal dominant optic atrophy
- LFSNHL, low frequency sensorineural hearing loss
- NEC, National Eye Clinic for the Visually Impaired
- OA, optic atrophy
- OGTT, oral glucose tolerance test
- WS, Wolfram syndrome
- wolframin
- WFS1
- autosomal dominant optic atrophy and sensorineural hearing impairment
- mutation analysis
Statistics from Altmetric.com
- ADOA, autosomal dominant optic atrophy
- LFSNHL, low frequency sensorineural hearing loss
- NEC, National Eye Clinic for the Visually Impaired
- OA, optic atrophy
- OGTT, oral glucose tolerance test
- WS, Wolfram syndrome
Hereditary optic neuropathy occurs with a prevalence between 1:10 000 and 1:50 000, and the most common type is autosomal dominant optic atrophy, ADOA (MIM #165500). This disease usually starts in childhood and is characterised by a progressive decline in visual acuity and temporal optic nerve pallor.1 Optic atrophy (OA) is genetically heterogeneous,2 and four autosomal loci have been identified: OPA1 on chromosome 3q28,3OPA3 on 19q13.2–q13.3.4OPA4 on chromosome 18q12.2–q12.3,5 and recently, OPA5 on chromosome 22q12.1–q13.1 in two unrelated French families with ADOA.6 In addition, juvenile optic atrophy is a key component of the autosomal recessive neurodegenerative disorder Wolfram syndrome (WS), caused by mutations in the WFS1 gene on chromosome 4p16.3.7
OPA1 seems most frequently involved in OA, and one particular mutation, c.2826delT, explains the high prevalence of ADOA, Kjer type, in Denmark.3,8OPA3 or Costeff optic atrophy syndrome (MIM #258501) is an autosomal recessive disorder associated with neurological abnormalities, and recently, mutations in the OPA3 gene were shown to segregate with ADOA associated with cataract.4OPA4 (MIM #605293) designates a locus for autosomal recessive optic atrophy, but the corresponding gene remains to be identified.5
ADOA associated with hearing impairment not linked to OPA1 has been described in several instances.9–15 One specific missense mutation, R445H, in OPA1 has been identified in four sporadic/familial cases with dual sensory impairment, and furthermore, in two families with ptosis, ophthalmoplegia, and movement disorders.16
Autosomal recessive WS is characterised by juvenile onset diabetes mellitus and optic atrophy, with 60% of the patients showing various degrees of hearing impairment by 20 years of age,17 and frequently endocrinological, psychiatric, urological, and neurological symptoms. Mutations in the WFS1 gene in WS are distributed over the entire coding region and result in loss of function of the encoded protein.18WFS1 has also been implicated in families with autosomal dominant isolated low frequency sensorineural hearing loss (LFSNHL) mapping to 4p16.7,19 The corresponding mutations are mainly missense mutations in exon 8, probably resulting in gain of function of the protein.20,21 Optic atrophy has not been reported as a concomitant feature in families with autosomal dominant low frequency hearing loss associated with WFS1 mutations. The fact that carriers of WFS1 mutations associated with WS do not have hearing impairment and heterozygosity for other WFS1 mutations resulting in non-syndromic autosomal dominant LSFHNL illustrates our incomplete knowledge of the function of the WFS1 protein and the consequences of the different types of WFS1 mutations.21
In this study, we performed extended linkage and mutation analyses in one Danish family with ADOA in combination with hearing impairment and identified the novel disease causing mutation.
METHODS
A three generation family (fig 1) was followed for 20 years at the National Eye Clinic for the Visually Impaired (NEC). Patient histories were recorded and the subjects underwent ophthalmological and optometric examinations at the NEC at regular intervals. Audiological and other medical information was also collected from hospital departments. The definitions used for the classification of hearing impairment are given by European Working Group on Genetics of Hearing Impairment.22 After an overnight fast, the members V:3, IV:4, and IV:6 underwent a standard 75 g oral glucose tolerance test (OGTT). Plasma glucose, serum specific insulin (excluding des(31, 32)– and intact proinsulin), serum C-peptide and HbA1C were analysed using standard methods (Steno Diabetes Center). The insulinogenic index was calculated as the 30 minute post-OGTT serum insulin (pmol/l) minus the fasting serum insulin (pmol/l) and divided by the 30 minute post-OGTT plasma glucose level (mmol/l). Results obtained from previous studies performed on age and sex matched glucose tolerant subjects at Steno Diabetes Center were used as control data. The investigations adhered to the tenets of the declaration of Helsinki and all participants gave informed consent for the investigations.

Five generation pedigree for family 148. The STS markers on chromosome 3, 4, and 18 each follow a haplotype map flanking candidate genes: D4S394 (WSF1), D3S3669 (OPA1), D18S450 (OPA4), and D22S275 (OPA5). An asterisk denotes STS marker results that exclude ADOA. STS marker D4S394 segregates together with the combined sensory affections in the members III:3, IV:4, IV:6, and V:3, and does not segregate together with the hearing impairment in III:5, III:7, IV:11, and IV:12. Individuals III:5 and III:7 had age related HI presenting after the age of 70 years (audiological data not shown) preferentially affecting the higher frequencies. V:4 had normal hearing. It was reported that II:2, born in 1881, who died at 78 years of age, had HI at old age. He was one of 10 siblings, of whom a sister (II:3) and a brother (II:4) had HI, as did as their father (I:2).
Blood samples were obtained from all available informative family members, and DNA was extracted from whole blood using standard procedures. DNA from 60 unrelated normal individuals from the Copenhagen Family Bank (families 604–1505)23 were used as controls. Ten closely linked polymorphic markers (D3S1601, D3S2418, D3S3669, D3S1523, D3S3642, D3S2305, D3S3562, D3S2748, and D3S1265) flanking the OPA1 gene on 3q28, four closely linked markers (D18S66, D18S450, D18S851, and D18S68) flanking OPA4 on 18q12.2–q12.3, and three closely linked markers (D4S412, D4S394, and D4S403) on 4p16.3, enclosing the WFS1 gene, were analysed. Furthermore, four closely linked markers (D13S1316, D13S175, D13S1275, and D13S787) flanking GJB3 and GJB6 on 13q12.11 were tested. One closely linked marker to OPA5, D22S275, was used to investigate for segregation with ADOA. All marker systems were typed by standard methods. Primers were radioactively end labelled, PCR was carried out under standard conditions and bands were separated by electrophoresis under denaturing conditions on polyacrylamide gels, and autoradiography was carried out.
Two point linkage analyses using the MLINK subroutine of FASTLINK package (version 5.1) were performed.24 Optic atrophy was modelled as a fully penetrant autosomal trait with a frequency of 0.001 and haplotype, and analyses were done for all chromosome regions.
PCR and sequencing primers were designed using the software Primer325 and intron located primers were designed to be a minimum of 100 bp from the intron−exon splice sites (table 1). Primers for sequencing of GJB6 exon 3, GJB2 exon 1 and exon 2,and PCR primers for detection of the 342 kbp (del-GJB6-D131830) large deletion of GJB626,27 are shown in table 1. Coding sequence exons in all four genes were sequenced in the affected people IV:4, IV:6, IV:11, and IV:12 (fig 1), and in addition IV: 6 was tested for the common Danish OPA1 founder mutation c.2826delT in exon 28.8
Primer information for PCR amplification and sequencing of WFS1
PCR was carried out under standard conditions according to the enzyme manufactures protocols (Taq DNA polymerase; Amersham Biosciences, Amersham, Bucks, UK). Reactions were carried out in 15 μl volumes containing the buffer, 2.5 μmol/l dNTP, 10 μmol/l of each primer, 0.008% cresol red (Sigma-Aldrich Co., St. Louis, MO, USA), 12% sucrose (w/v), and 50–100 ng templates DNA. Standard reaction conditions for all primers were: 95°C for 5 minutes; 40 cycles of 95°C for 30 seconds, 56.4°C for 30 seconds and 72°C for 1 minute; followed by 5 minutes at 72°C. PCR reactions were separated in a 2% agarose gel and stained with ethidium bromide in 1× TBE buffer before sequencing. Both strands were sequenced directly using the PCR primers and a commercial kit (BigDye version 1.0, Applied Biosystems, Foster City, CA, USA) without further modifications and run on an ABI 370 sequencer (Applied Biosystems). Sequence data were analysed using standard software (Chromas, version 2.1; Australia) and sequence alignments were carried out using ClustalW28 or BLAT29 respectively. All exons were completely sequenced for two affected people, IV:6 and IV:12. PCR products of WFS1 exons 1, 2, 5, 8-2, 8-3, and 8-4 were generated with Platinum Taq DNA polymerase (Invitrogen A/S, Denmark) using 1.5 mmol/l Mg2+ under the same PCR conditions as above. Diagnostic restriction enzyme digest of the mutation c.2590G→A was carried out by PCR of exon 8 resulting in amplification of a 783 bp PCR product using primer pair 8:4F and 8:4R (table 1). Digestion with the restriction enzyme Bsp1286I resulted in four DNA fragments (69, 177, 218, and 319 bp), representing the wild type allele, and one additional 246 bp fragment (69 plus 177 bp) due to the mutation in one Bsp1286I site in affected heterozygotes. The digested PCR products were separated by 2% agarose gel electrophoresis with 1×TBE buffer.
RESULTS
The main clinical characteristics (table 2), and the audiometric pattern and course of the hearing impairment are shown (fig 2, table 3). An OGTT showed that IV:6 had undiagnosed diabetes, IV:4 had impaired glucose tolerance, and V:3 had normal glucose tolerance according to the WHO 1999 definition. Furthermore, IV:6 and V:3 had poor pancreatic β-cell function as evaluated by the insulinogenic index (0.7 and 2.1, respectively, compared to age and sex matched glucose tolerant control subjects; reference insulinogenic index: mean (SD) males 27.0 (11.2), females 25.0 (13.5)).
Clinical characteristics, audiometric pattern, and course of the hearing impairment in patients
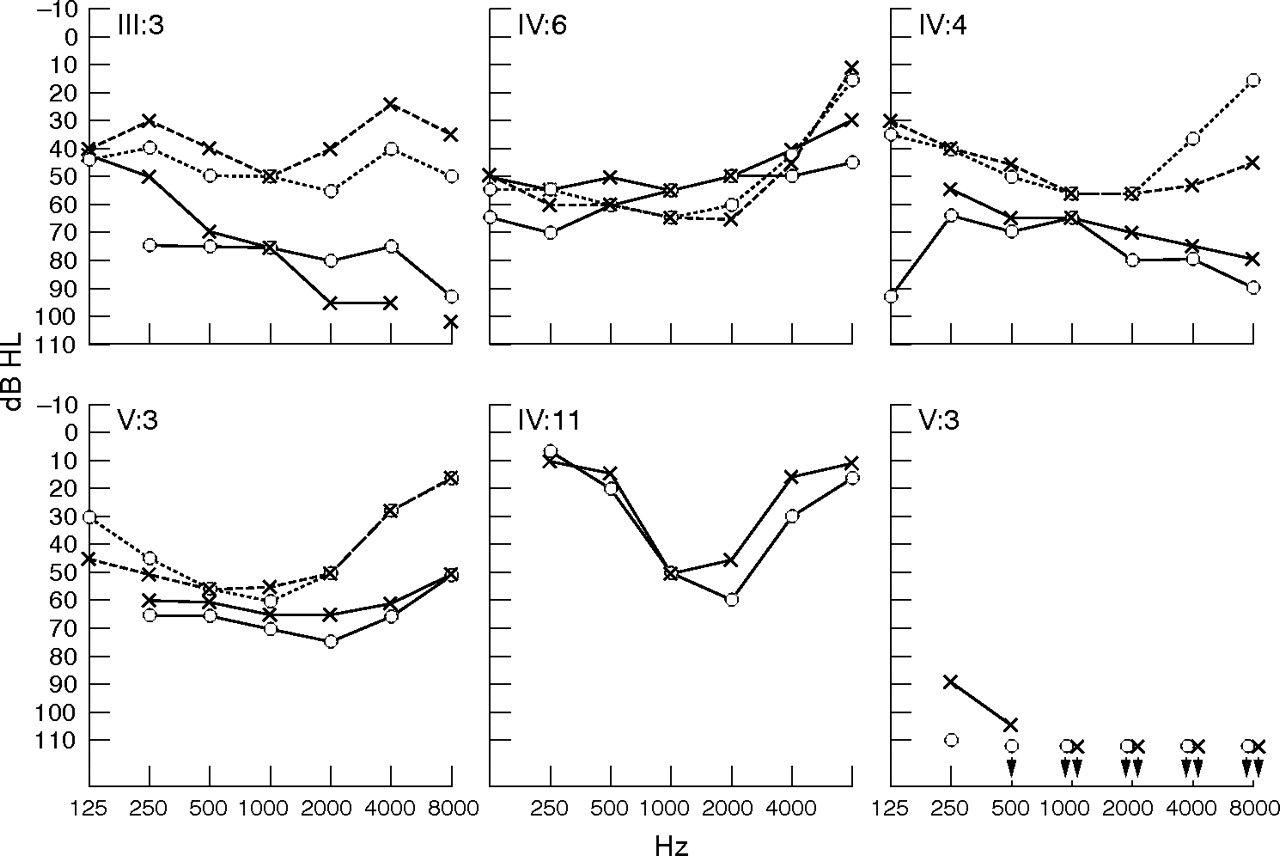
Audiograms showing the six affected people in family 148. Four members (III:3, IV:4, IV:6, and V:3) are heterozygous for the WFS1 mutation and two (IV:11 and IV:12) are homozygous normal. Broken lines indicate the first examination and solid lines the latest examination (age given below each audiogram). Circles and crosses represent air conduction thresholds for the right and left ear, respectively. Audiological information about mutation carriers is summarised in the text (table 2). The audiograms from the WFS1 mutation carriers are described (table 2), and in all instances affect the low frequencies. IV:11 (male, aged 55 years) had a deeply U-shaped audiogram and progressive HI, and used a hearing aid from 11 years of age. IV:12 (female, aged 44 years) had congenital profound HI and used sign language. She also had cleft palate.
Haplotype analyses with markers flanking the genes OPA1, OPA4, OPA5, GJB2, and GJB6 did not support those loci on chromosomes 3q28, 18q12.2–q12.3 and 22q12.1–q13.1, because of lack of co-segregation with ADOA and negative LOD scores for close linkage (fig 1, data not shown). Marker D4S394 close to the WFS1 gene on 4p16.3 gave a positive LOD score (Z = 1.61, θ = 0.0) for this area.
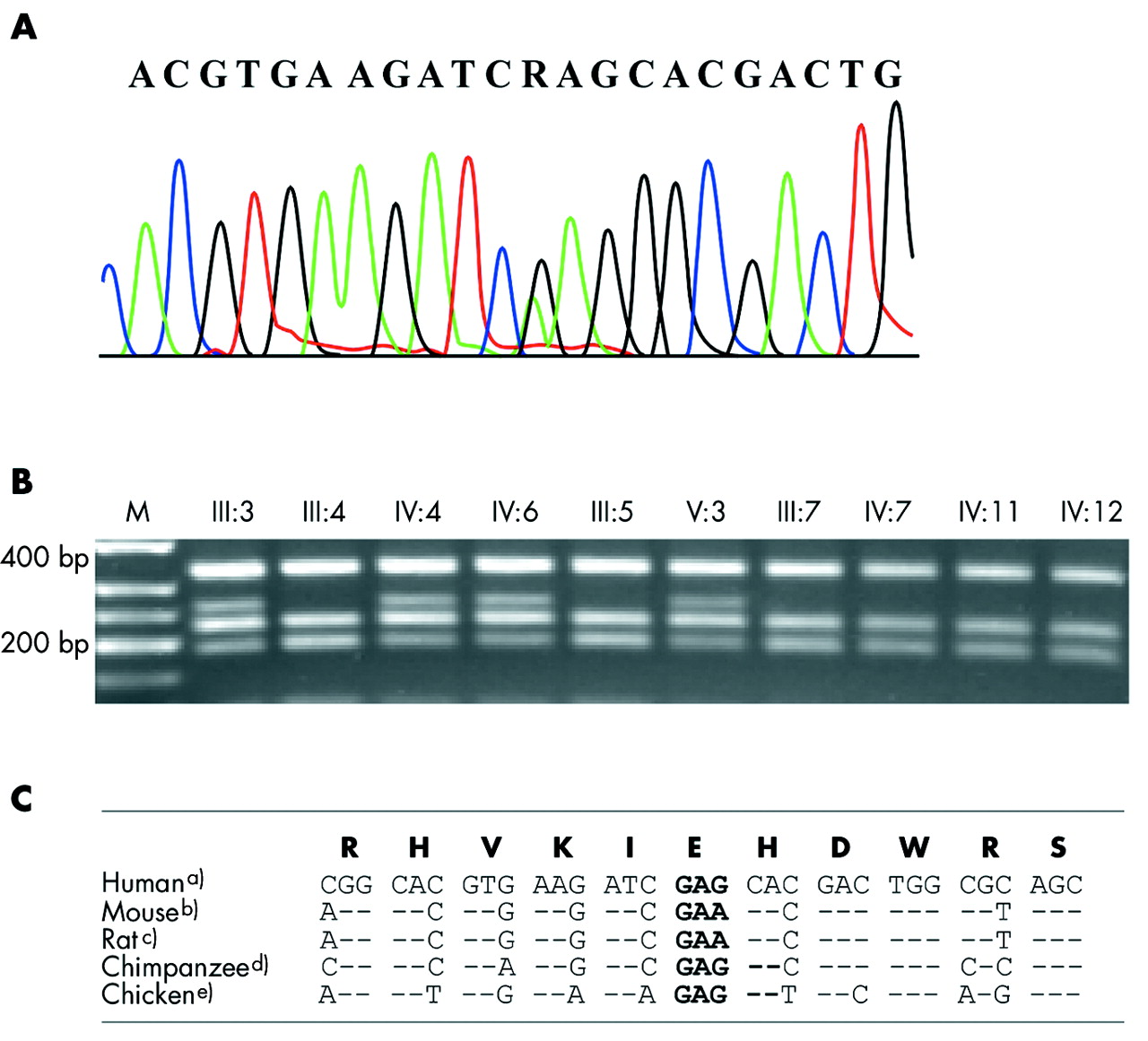
Mutation analysis of all coding exons in OPA3, GJB2, and GJB6 in IV:6, IV:11, and IV:12 gave negative results. The gene OPA1 did not have the common Danish founder mutation c.2826delT (frameshift mutation in exon 28) in IV:6 and IV:12. The OPA3 gene was excluded based on sequencing in IV:6 and additionally, the single nucleotide polymorphism rs3826860 in exon 2 found in IV:6 showed heterozygosity, which excluded the presence of a large deletion of the gene. Mutation analysis of exons 1–8 of the WFS1 gene in IV:6, IV:11, and IV:12 revealed one novel missense mutation (c.2590G→A in exon 8, only in person IV:6; fig 3A).

{kind=link}
{kind=link}
{kind=link}
(A) Sequencing chromatogram of the mutation c.2590 G→A in IV:6. The affected person is heterozygous for the missense mutation E864K and the G→A substitution is denoted by an "R". (B) The mutation segregates with sensorineural hearing loss in family #148. The 783 bp PCR product digested by Bsp1286I presents a 246 bp fragment (69 plus 177 bp) in heterozygotes in addition to the wild type fragments (69, 177, 218, and 319 bp) after separation in 2% agarose gel. Roman numbers above the gel refer to the family pedigree (fig 1). M is the DNA ladder (150, 200, 250, 300 and 400 bp) (C) Alignment of the 11 amino acid codons around the E864K mutation demonstrate conservation of the protein sequence and redundancy in the DNA sequence (sequence data: UCSC Genome Browser;34 (a) Homo sapiens chr4:6368639–6368671, NCBI build 34 July 2003; (b) Mus musculus chr5:35260341–35260373, NCBI build 30 February 2003; (c) Rattus norvegicus chr14:79406244–79406276, HGSC version3.1, Jun 2003; (d) Pan troglodytes chr3:6485955–6485987, NCBI build 1, version 1, November 2003, (e) Gallus gallus chr4:79834614–79834646, galGal, February 2004).
The disease phenotype segregated with this WFS1 mutation demonstrated by the restriction enzyme Bsp1286I digest of the PCR amplicons (fig 3B) and the mutation was absent in unaffected family members and in those with isolated hearing impairment (IV:11 and IV:12). The WFS1 mutation was not found among 60 normal control individuals.
DISCUSSION
A Danish family with juvenile onset of optic atrophy and associated hearing impairment was investigated. Haplotype and sequence analysis excluded mutations in OPA1, OPA3, OPA4, OPA5, GJB2, and GJB6. A positive LOD score of Z = 1.61 for 4p16.3 followed by sequencing of the WFS1 gene led to identification of a novel WFS1 missense mutation, E864K.
Four individuals carrying this mutation had optic atrophy and hearing impairment. The three with the most extended observation periods (III:3, IV:4, V:3) presented initially with a pure sensorineural moderate hearing impairment with shallow mid frequency U-shaped audiograms (fig 2, tables 2 and 3). The hearing impairment was progressive, becoming moderate to severe with flat or gently sloping audiogram configurations. The hearing impairment found in other family members was phenotypically different. In III:5 and III:7, the hearing impairment was age related, presenting in old age. The profound congenital hearing impairment in IV:12 and the more moderate deeply U-shaped hearing loss in her brothe (IV:11) are so far unexplained, as we excluded mutations in the coding regions of GJB2, and GJB6.
The type of hearing impairment in V:3, IV:6, IV:4, and III:3 significantly affected the lower frequencies, although only in IV:6 did they fulfill the stringent definition of LFSNHL.
The metabolic examinations of three individuals with the WFS1 mutation revealed that two of them had overt but undiagnosed diabetes (IV:6) or impaired glucose tolerance (IV:4) and that two mutation carriers (IV:6 and V:3) had impaired insulinogenic index. Thus, these data of impaired glucose regulation indicate that the WFS1 mutation may affect the pancreatic β-cell function as well.
Altogether, at least 20 different missense mutations in WFS1 have been reported to associate with autosomal dominant LFSNHL; 19 in exon 8 and 1 in exon 5.30,31 Seventeen cluster in the C-terminus of the WFS1 protein, one is located in the transmembrane domain 9, one in the cytoplamatic domain 5, and one sporadic case in the N-terminus.20,21,32,33 It is well known that mutations in the WSF1 gene can cause variable expression, giving rise to different clinical complications such as hearing, vision, diabetes, and depression. In a large non-syndromic autosomal dominant LFSNHL family reported by Young et al21, one individual who was homozygous for a A716T mutation in the C-terminus had partial WS features (diabetes mellitus from 3 years of age, 20–30 dB HL in the frequency range 0.5–4 kHz and cataract, but normal renal ultrasound and no optic atrophy).21 These findings clearly demonstrate that access to in vitro functional tests of the WFS1 protein is crucially wanted in order to assess the implications of the individual mutations.
In conclusion, this is the first report of a missense mutation (c.2590G→A, E864K) in the WFS1 gene in a family with ADOA in combination with hearing impairment. This mutation changes the C-terminus of the protein by substitution of the non-charged amino acid glutamine by the positively charged lysine and is predicted to causes a critical change of the part of the WFS1 protein that is at the endoplasmic reticulum lumen. The glutamine E864 is conserved in the WFS1 protein in human, mouse, rat, chimpanzee, and chicken (fig 3C), supporting our interpretation that the mutation is causative. Our findings illustrate that mutations in WFS1 may be associated with a broader clinical spectrum of phenotypes than previously reported, in terms of ADOA, impaired glucose regulation, and audiological characteristics. The increasing recognition of different inheritance patterns and the spectrum of a broad clinical presentation associated with mutations in the OPA1, OPA3, and WFS1 genes are striking. Such observations warrant a strong awareness of the possibility of multiple organs being affected in patients seen by both ophthalmologists and audiologists. Through meticulous characterisation of the clinical implications of mutations in these genes, we may obtain a deeper understanding of the normal biological functions of WSF1 as well as other candidate genes involved in hearing and vision disorders.
Acknowledgments
We thank all family members for their participation in this study. A Mikkelsen, L Rasmussen. and M Christensen are thanked for excellent technical assistance, and K Kjær for helpful discussion. This work was supported by the Danish Eye Research Foundation (Øjenfonden), Værn om Synet and the Danish Research Council, La Fountaine Foundation, the Novo Nordic Foundation, the Danish Medical Research Council, the Danish Diabetes association, the Oticon Foundation, and Widex A/S.
REFERENCES
Footnotes
-
Competing interests: there are no competing interests.