Article Text

Abstract
Background: Genetic testing for hereditary cancer syndromes contributes to the medical management of patients who may be at increased risk of one or more cancers. BRCA1 and BRCA2 testing for hereditary breast and ovarian cancer is one such widely used test. However, clinical testing methods with high sensitivity for deleterious mutations in these genes also detect many unclassified variants, primarily missense substitutions.
Methods: We developed an extension of the Grantham difference, called A-GVGD, to score missense substitutions against the range of variation present at their position in a multiple sequence alignment. Combining two methods, co-occurrence of unclassified variants with clearly deleterious mutations and A-GVGD, we analysed most of the missense substitutions observed in BRCA1.
Results: A-GVGD was able to resolve known neutral and deleterious missense substitutions into distinct sets. Additionally, eight previously unclassified BRCA1 missense substitutions observed in trans with one or more deleterious mutations, and within the cross-species range of variation observed at their position in the protein, are now classified as neutral.
Discussion: The methods combined here can classify as neutral about 50% of missense substitutions that have been observed with two or more clearly deleterious mutations. Furthermore, odds ratios estimated for sets of substitutions grouped by A-GVGD scores are consistent with the hypothesis that most unclassified substitutions that are within the cross-species range of variation at their position in BRCA1 are also neutral. For most of these, clinical reclassification will require integrated application of other methods such as pooled family histories, segregation analysis, or validated functional assay.
- A-GVGD
- BRCA1
- co-occurrence
- Grantham difference
- missense substitution
- polymorphism
Statistics from Altmetric.com
Genetic testing for hereditary cancer syndromes is increasingly contributing to the medical management of patients who may be at markedly increased risk of one or more cancers. Testing of a patient who has a strong family history will ideally result in the discovery of a clearly deleterious cancer predisposing mutation, leading to options such as aggressive screening, prophylactic surgery, or chemopreventive strategies. Unfortunately, in many cases where no clearly deleterious mutation is found, a sequence variant of uncertain clinical significance, most often a missense substitution, is found. In such cases, patients, and the healthcare providers who counsel them, are left with ambiguous test results that are of little help in determining appropriate cancer risk-reduction strategies.
Germline loss of function mutations in BRCA1 and BRCA2 confer high risk of breast cancer and ovarian cancer and confer elevated risks of a number of other cancers.1–5 Testing for mutations in these genes has become one of the most widely used hereditary cancer tests, with over 70 000 patients tested to date. Although 13.5% of patients tested through full sequence analyses of both BRCA1 and BRCA2 at Myriad Genetic Laboratories (MGL) are found to carry a deleterious mutation, 12% of patients who do not carry a clearly deleterious variant are found to carry an uncertain variant (database query updated from that of Frank et al6).
Recently, Goldgar et al7 developed a method for analysis of unclassified missense substitutions in BRCA1 and BRCA2 that integrates four types of data: segregation of sequence variants of interest in pedigrees; pooled family histories of index cases who carry the variant versus all index cases tested; co-occurrence of the variant with clearly deleterious variants in the same gene; and cross-species protein multiple sequence alignment followed by comparison of the physico-chemical characteristics of the amino acids observed at the point of the mutation (Grantham analysis). Each of these data types has its strengths and weaknesses. Segregation analysis and pooled family history analysis are both pure human genetics measures; they provide fairly direct measures of disease susceptibility. However, both require accurate family history data, which may be difficult to obtain. Co-occurrence of variants of interest with clearly deleterious mutations takes advantage of the highly penetrant embryonic lethal phenotype conferred by most BRCA1-null genotypes8,9,10,11,12,13,14 and is thus both a human genetics measure and a type of functional assay, but one in which genetics and function are measured indirectly. Sequence alignments and Grantham analyses are measures of evolutionary fitness that are only indirectly tied to disease susceptibility. In contrast to segregation analysis and summary family history, co-occurrence data can be obtained directly from systematically compiled mutation screening databases. Similarly, sequence alignment/Grantham analysis can readily be applied to any observed missense substitution.
One strength of the integrated method is that each of the four types of data analysis that it has integrated was developed as an independent estimator of the likelihood that a sequence variant confers a high cancer risk versus being neutral or of little clinical significance (neutral/LCS). Likelihood calculations from the four methods are multiplied to reach a final result. One does not need to use all four types of data to analyse any particular variant; analyses using two or three data types are also perfectly valid. In the following study we have used co-occurrence data and a modified approach to sequence alignment/Grantham analysis to look at all 452 missense substitutions observed in BRCA1 in a series of 40 000 full-sequence BRCA1 and BRCA2 tests conducted at MGL (B1&2 40K set), with the goal of identifying missense substitutions that are neutral/LCS.
METHODS
BRCA1 and BRCA2 testing (BRACAnalysis®)
Full sequence analyses of BRCA1 and BRCA2 were performed by direct gene sequencing as previously described.15 In order for a test to be performed, the test request form must be completed by the ordering healthcare provider and the form must be signed by an appropriate individual indicating that “informed consent has been signed and is on file”. Patient samples were each assigned a unique bar code for robotic specimen tracking. Most samples were received as 7 ml of anti-coagulated blood, from which DNA was extracted and purified from leukocytes isolated from each sample. Aliquots of patient DNA were each subjected to polymerase chain reaction (PCR) amplification. The amplified products were each directly sequenced in the forward and reverse directions using fluorescent dye-labelled sequencing primers. Chromatographic tracings of each amplicon were analysed by MGL’s sequence analysis software followed by visual inspection and confirmation, assisted by comparison of the proband sequence to a consensus wild type sequence constructed for each amplicon. Each genetic variant (exclusive of non-reportable polymorphisms) was independently confirmed by repeated analysis including PCR amplification of the indicated gene region(s) and sequence determination.
All mutations and genetic variants were named according to the convention of Beaudet and Tsui,16 and all of them have been submitted to the BIC database at http://research.nhgri.nih.gov/bic/. Nucleotide numbering starts at the first transcribed base of BRCA1 according to GenBank entry U14680. (Under this convention, the mutation commonly referred to as “185delAG” is named “187delAG”. However, in this paper, we refer to this variant by its more common name.)
Co-occurrence analysis
The expression for using co-occurrence data to calculate the likelihood that a variant is deleterious versus neutral/LCS was developed in Goldgar et al.7 Briefly, if the sequence variant of interest was observed n times, k of which were in individuals who also carry a clearly deleterious variant, the appropriate binomial likelihood ratio is:

where p1 is the probability that an individual in the test population who carries an unclassified neutral variant also carries (in trans) a deleterious mutation, and p2 is the probability that an individual in the test population who carries an unclassified deleterious variant also carries (in trans) a deleterious mutation. The overall frequency of clearly deleterious BRCA1 mutations found by BRACAnalysis in the B1&2 40K set was 8.1%, and we take p1 to be 50% of that frequency. BRCA1 homozygote and compound heterozygote genotypes are quite likely embryonic lethal and consequently extremely rare; accordingly, we have set p2 = 0.0001 for these calculations.7
In addition, our basic query of the BRACAnalysis database gave us the identity of every missense substitution observed, the number of times each was observed, the number of times each was observed with a clearly deleterious mutation in BRCA1, the number of different clearly deleterious mutations with which each was observed, and the number of times each was observed with a clearly deleterious mutation in BRCA2. We were also able to query the co-occurring deleterious mutations to see how often they were seen independently of the missense substitution of interest. In our analysis of unclassified missense substitutions, only co-occurrences with independently observed deleterious mutations were used in the likelihood calculation.
Haplotype analysis
Using 14 common polymorphisms in BRCA1 that are within the sequences covered by BRACAnalysis (exon 4 –49 C>T, IVS8-58delT, Q356R, D693N, S694S, L771L, P871L, E1038G, S1040N, K1183R, R1347G, S1436S, S1613G, and M1652I), we have defined the 10 most common haplotypes in our test population (Hendrickson et al17 and T Scholl, manuscript in preparation). Due to the simple haplotype structure of BRCA1, no two genotypes that result from pairs of these haplotypes are identical. All the sequence variants that we needed to analyse, both the unclassified substitutions and the deleterious variants with which they co-occurred, were examined for the haplotype contexts in which they were seen. For sequence variants that were observed more than approximately five times, this usually resulted in a single unambiguous haplotype assignment. Once variants had been assigned to specific haplotypes, we looked at the genotypes of the patients in whom co-occurrences were observed. In those cases where we were able to determine haplotypes for both the mutation and the unclassified substitution, it was sometimes clear that the test subject was a heterozygote for the two haplotypes and that the two variants were therefore in trans.
Sequencing of Monodelphis BRCA1
Peptide sequences of the individual coding exons of human BRCA1 were searched against genomic sequence reads from the Monodelphis domestica genome sequencing project by tBLASTn.18 Most exons of the M domestica BRCA1 ortholog were identified with little ambiguity. PCR and sequencing primers were designed based on the predicted exon sequences; primers were selected so that predicted PCR product lengths would be between 1 and 2 kb, missing exons in the assembly would be spanned, and products would overlap to allow complete sequencing. cDNA was prepared from colon and testis samples of two individual opossums. After PCR, products were gel purified and sequenced with BigDye dye terminator chemistry. Our sequence, which has 47% amino acid sequence identity to human BRCA1, has been submitted to GenBank under accession no. AY994160 and also used in the alignment described below.
Creation and analysis of the multiple sequence alignment
The BRCA1 protein multiple sequence alignment used for this analysis contained 12 full-length BRCA1 sequences. The evolutionary relationships and % sequence identities between most of these sequences were described previously.19 The multiple sequence alignment was made with the alignment program 3DCoffee, which also incorporates alignment to x ray and NMR structures.20 3DCoffee was run using Mlalign_id_pair, Mslow_pair, and Mclustalw_aln to generate amino acid alignments and Mfugue_pair to generate structure sequence alignments. GenBank accession numbers for BRCA1 protein sequences used in the alignment were as follows: human, NP_009225; chimpanzee, AAG43492; gorilla, AAT44835; orangutan, AAT44834; rhesus macaque, AAT44833; mouse, AAD00168; dog, AAC48663; cow NP_848668; opossum, AAX92675; chicken, NP_989500; Xenopus, AAL13037; and Tetraodon, AAR89523. For the structure component of the alignment, we used BRCA1 RING NMR structure 1JM7.pdb and the BRCA1 BRCT repeat crystal structures 1JNX.pdb and 1T29.pdb.21–23 A parsimony based method was used to calculate the minimum number of missense substitutions required to create the observed alignment, taking into account the underlying phylogenetic tree.19,24 Because we are interested in human disease genetics, our subsequent analyses only considered sequence variation at positions in the alignment where the human sequence has a residue. On the other hand, absence of an amino acid in the alignment of a non-human BRCA1 at a position where the human sequence does have a residue was considered a sequence variation.
The number of slowly substituting positions (class 2 or SS), fast substituting positions (class 3 or FS), and the relative odds that a position in BRCA1 where 0, 1, 2, … n substitutions are observed is either SS or FS, were calculated from the protein multiple sequence alignment using the modified Fitch covarion model “Model 3” of Abkevich et al.19,24 The relative odds that a position in BRCA1 is either SS or FS is identical to the sequence conservation likelihood ratio used in Goldgar et al.7
Grantham analysis
For two amino acids i and j with sidechain compositions Ci and Cj, polarities Pi and Pj, and volumes Vi and Vj, the standard Grantham difference formula is:
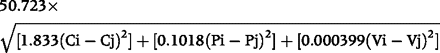
C, P, and V values for the 20 common amino acids are given with Grantham’s definition of the measure.25
The set of amino acids observed at a particular position in a protein multiple sequence alignment will have minimum and maximum values of C, P, and V. For calculation of the Grantham variation (GV) of a position, Cmax replaces Ci, Cmin replaces Cj, and so on, in eq 1. A sample calculation of GV is given in fig 1C. Difficulties arise at gaps in the sequence alignment. We arrange our alignments so that the first sequence is the human sequence and successive sequences are from species that are successively more distantly related to humans. GVs are calculated sequentially from closely related species to distantly related species. At the first appearance of a gap at a particular position in the alignment, we set Cmin and Cmax to 0 and 3, respectively (the highest value of C for an amino acid is 2.75); we use Pmin and Pmax from the observed amino acids; and we set Vmin to 0 but set Vmax to the largest V from the observed amino acids. If the position is again gapped in more distantly related species, and if the positions immediately before and immediately after the position of interest are also gapped, then we set all Cmin, Pmin, and Vmin to 0 and Cmax, Pmax, and Vmax to 3, 14, and 175 (which are all slightly above the highest values for normal amino acids).

Sample calculation of GV and GD using M1652I as an example. (A) Thirty amino acid sequence alignment from the beginning of the BRCA1 BRCT domain. Position 1652 is marked in grey. (B) For each amino acid aligned to human position 1652, we give the amino acid sidechain composition (C), polarity (P), and volume (V). Starting with human and working sequentially to the Tetraodon nigroviridis sequence, we determine the minimum and maximum observed values of C, P, and V; these are used to calculate GV according to eq 2. Starting with human and working sequentially to the T nigroviridis sequence, we then determine the difference between C, P, and V for the missense variant (Ile in this case; C, P, and V values for Ile are marked in grey) and the range of variation observed to that point in the alignment. The differences, given in the columns headed ΔC, ΔP, and ΔV, are used to calculate GD according to eq 2.
Each missense substitution is characterised by values Cm, Pm, and Vm. These are either below, within, or above the range of variation captured in the GV of the position of interest. For calculation of the Grantham deviation (GD), if Cm<Cmin, then the C component of eq 1 is replaced by (Cmin−Cm); if Cmin⩽C⩽Cmax, then C component of eq 1 = 0; if Cm>Cmax, then the C component of eq 1 is replaced by (Cm−Cmax). Corresponding substitutions are made for the P and V components of eq 1. A sample calculation of GD is given in fig 1D. The overall method combining sequence alignment with calculation of GV and GD is called A-GVGD.
Statistical methods
Calculations of GV and GD were implemented in APL (APLX Version 2.0.9, MicroAPL, Uckfield, East Sussex, UK). The BRCA1 with BRCA2 ascertainment for BRACAnalysis odds ratios was calculated in a series of contingency tables. For the disease status axis of these tables, “controls” are individuals who carry a clearly deleterious mutation in BRCA2, and “cases” are individuals who were not found to carry a deleterious BRCA2 mutation. Categories on the genotype axis are pooled sets of sequence variants that meet specific selection criteria, as described in the legend to fig 3. Confidence intervals were estimated using Miettinen’s test-based approximation.
RESULTS
Co-occurrence analysis
The idea of using co-occurrence of clearly deleterious sequence variants with unclassified missense substitutions to classify missense substitutions in BRCA1 arises from two independent sources. One is a series of mouse and Xenopus studies which show that complete loss of BRCA1 function confers a highly penetrant embryonic lethal phenotype.8,9,10,11,12,13,14 The second is observation of a clear deficit of Ashkenazi BRCA1 homozygotes/compound heterozygotes in MGL’s BRACAnalysis database.6,19 Of the 452 missense substitutions in the B1&2 40K set, 72 have been observed in an individual who also carried a clearly deleterious BRCA1 mutation. Fifteen of the 16 missense substitutions that currently are classified as neutral/LCS fall into this group, as do 57 unclassified missense substitutions (table 1). None of the 28 missense substitutions that are currently classified by MGL as deleterious or favour deleterious were observed in a patient who carries another clearly deleterious mutation in BRCA1.
Observations of missense substitutions in BRCA1 in individuals who also carry a clearly deleterious BRCA1 mutation
At a superficial level of analysis, the main pitfall of co-occurrence data is that, for purposes of classification of missense substitutions, an observation of co-occurrence is only meaningful if the unclassified missense substitution and the clearly deleterious mutation are in trans. Fortunately, as a consequence of full sequence testing, we have additional data that can help to discriminate cases of cis from trans co-occurrence. First, in addition to the number of times that each missense substitution has been observed with a clearly deleterious mutation, we also know the number of different deleterious mutations with which it was observed to co-occur and their individual identities. Second, for each deleterious variant that has been observed in a patient who also carried an unclassified substitution of interest, we know the number of times that the deleterious variant was observed with and without the substitution of interest. Combined, these data allow us to identify substitutions that actually are in cis with a clearly deleterious mutation. Third, for recurrent sequence variants we can usually determine, using single nucleotide polymorphisms (SNPs), independent of any disease consideration, the common haplotype on which that variant is found.
Taking advantage of these additional data, we can add four layers of caution to our calculation of the likelihood ratio using co-occurrence data (cooc-LR). (i) When the deleterious variant involved in a co-occurrence has been observed only once, we do not know whether the unclassified substitution and the deleterious variant are independent. For application of cooc-LR to the unclassified variants, these are ignored. (ii) When the unclassified variant and deleterious variant are not usually observed independently, co-occurrences of this pair of variants are ignored. (iii) If these precautionary subtractions leave two or more distinct co-occurrences, we can assume that at least n−1 of them are in trans; this point is explained further in the next paragraph. (iv) If these precautionary subtractions leave only one distinct co-occurrence, we use our knowledge of SNP-based haplotypes to determine whether the individual who carries the two variants of interest has a SNP genotype that is compatible with being a heterozygote for the SNP haplotypes on which the unclassified missense substitution and the deleterious mutation are usually observed and incompatible with being a homozygote for either of those haplotypes. Meeting this condition confirms a trans co-occurrence; the confirmed trans co-occurrence(s) is then used in the calculation.
When an individual is observed to carry two rare sequence variants, in the same gene, that usually segregate independently, the most likely explanation by far is that the two sequence variants have been inherited in trans. However, there can be exceptions. One possibility is that the subject could have inherited a very rare recombinant chromosome that carries the two sequence variants in cis. In this scenario, for BRCA1, it is extremely unlikely that both sequence variants are deleterious. This is because the recombination that brought the two sequence variants into cis would have to have taken place in an ancestor who inherited the two sequence variants in trans. But if both variants are deleterious, that ancestor’s genotype would have been highly penetrant embryonic lethal,8,9,10,11,12,13 and, if not lethal, would have interfered directly with the process of recombination.14 An alternative possibility is that the subject could have inherited a chromosome on which the second of the two sequence variants is a new mutation that happens to be identical to the rare sequence variant that normally segregates independently of the first variant (or vice versa). However, excepting substitutions at CpG dinucleotides and length variations in repeated sequence elements, multiple independent origins of the same human sequence variant are quite rare. Thus the probability that we will have observed a BRCA1 allele bearing one of the relatively rare unclassified missense substitutions of interest that has been hit by the second origin of a specific deleterious BRCA1 mutation is low, and the probability that we will observe an unclassified missense substitution-bearing allele that has been hit twice by second origins of deleterious mutations is infinitesimal. This logic also holds for the occurrence of a second independent origin of one of the unclassified missense substitutions of interest on a deleterious mutation-bearing BRCA1 allele. Thus, for rare sequence variants, we can assume that at most one observation of co-occurrence with an independently observed deleterious mutation is due to occult co-occurrence in cis.
We applied this analysis to the 33 missense substitutions that were observed to co-occur with a deleterious variant two or more times. Of these, 15 are classified as neutral/LCS, while 18 are currently unclassified. cooc-LRs for all 15 known neutral/LCS variants were below 1×10−2 (table 2A). The neutral substitution with the fewest co-occurrences was Y856H (observed once with each of two different deleterious mutations). Even though we ignored one of its two co-occurrences in the calculation, its cooc-LR was below 0.01.
Missense substitutions in BRCA1 that have been observed with two or more known deleterious mutations
Following the cautious approach outlined above, cooc-LRs were also calculated for the 18 unclassified missense substitutions that were nominally observed to co-occur two or more times (table 2B). Although not strictly necessary, a haplotype based cis-trans test was made for at least one double carrier of each of these substitutions. Seven of the substitutions were observed with two or more different, independently segregating deleterious mutations; cooc-LRs for these were all below 1×10−2. One of these substitutions, D1546N, was observed once each with two different deleterious mutations. Asn1546 and the two deleterious mutations were all on the same SNP haplotype, rendering the cis-trans test uninformative. However, because two independently segregating deleterious mutations were involved in these co-occurrences, we conclude that at least one of these, and probably both, are bona fide trans co-occurrences. Five of the substitutions were observed with only one independently segregating deleterious mutation. In each case, BRCA1 SNP genotypes for the double carriers were as expected for a trans-carrier of the two variants of interest, resulting in cooc-LRs for these five substitutions of 1.2×10−2 or less. The result with M1008I was particularly interesting. Ile1008 was observed 103 times; three of these observations were with a clearly deleterious mutation, but always the same mutation. Superficially, one might expect from this pattern that a rare mutation had occurred on an Ile1008 chromosome and that the co-occurrences would be in cis. However, Ile1008 is most often observed in individuals of Ashkenazi ancestry. The mutation with which it has been observed three times is 185delAG. Among Ashkenazim, the frequency of 185delAG is higher than the summed frequency of all other clearly deleterious mutations in the gene; consequently, it is not surprising to have observed three individuals who carry this pair of variants in trans. Finally, six of the unclassified missense substitutions were observed with a deleterious mutation more than once, but always with the same deleterious mutation and in observation patterns best explained by co-occurrences in cis.
Measures of evolutionary variation and observed deviation
The basic logic behind the use of protein multiple sequence alignments to identify missense substitutions that are likely to be either neutral/LCS or deleterious breaks down into two components: (i) missense substitutions at positions that are highly functionally constrained tend to alter protein function while substitutions at positions that are not so constrained are less likely to alter function, and (ii) missense substitutions that are outside the range of variation that is evolutionarily tolerated at their position in the protein tend to alter protein function whereas those that are within the range of variation tend to have little effect on protein function. Previously, we have used two different approaches to this problem.7,19 The approach taken in Abkevich et al19 made only qualitative use of evidence that a position in the BRCA1 was functionally constrained or not and then made quantitative use of the fit between an observed missense substitution and the range of variation at its corresponding position in a BRCA1 protein multiple sequence alignment. Neither component of that analysis was formatted as a likelihood ratio; consequently, that approach shared with SIFT and PolyPhen27,28 the flaw that it is not easily integrated into a proper multi-model likelihood calculation. The approach taken in Goldgar et al7 decomposed alignment/Grantham analysis into two likelihood expressions: the first was based on the ratio of probabilities that the position at which a missense substitution is observed is functionally constrained or not, and the second was based on the Grantham difference25 for the missense substitution versus the canonical human residue at its position in BRCA1. The first component, which we shall refer to as the constrained position likelihood ratio (con-LR), in agreement with Abkevich et al,19 appeared to have good predictive power. However, the way that the Grantham differences were used in the second component of the analysis did not correlate well with the other measures used in that work. This was probably because Grantham differences, used by themselves, do not make use of the fit between an observed missense substitution and the range of variation at its corresponding position in a BRCA1 protein multiple sequence alignment.
The fundamental problem is that the standard Grantham difference is a pairwise comparison pressed into service for an application that would benefit from a genuine multiple comparison. As formulated in Goldgar et al,7 Grantham differences are being used in a simple pairwise format. As formulated in Abkevich et al19 and also in the recently proposed Grantham ratio,29 the Grantham approach is extended to a multiple pairwise comparison; this is better but not entirely satisfactory. In order to achieve a true simultaneous multiple comparison, we introduce the Grantham variation (GV) and the Grantham deviation (GD) scores as follows.
In a 3-space where the axes are measures of amino acid sidechain composition (C), polarity (P), and volume (V), the original Grantham difference for a pair of amino acids is the Euclidean distance between the C, P, and V values for those two amino acids (with scaling constants applied to squared C, P, and V differences).25 For the amino acids observed at a specific position in a protein multiple sequence alignment, we consider the smallest rectangular box that contains all the corresponding points in Grantham space. The GV is then the length of the longest diagonal of the box. Given the point in Grantham space corresponding to the amino acid caused by a missense substitution, we define the GD as the shortest distance from that point to the bounding box. Any point inside the box has GD = 0 (see Methods for exact formulas). Consequently, using the same scale as Grantham differences, these two measures provide a numerically precise yet quite natural description of the magnitude of sequence variation at any position in a protein multiple sequence alignment, and the fit between any given human missense substitution and the sequence variation present at its position in a multiple sequence alignment.
Characterisation of the multiple sequence alignment and the Grantham deviation
Calculations of GV, GD, and the con-LR for the 452 missense substitutions observed in the B1&2 40K set are based on a 12-sequence alignment that contains full length BRCA1 sequences from eight placental mammals plus M domestica (gray, short-tailed opossum), G gallus (chicken), X laevis (African clawed frog), and T nigroviridis (green-spotted pufferfish). The sequence alignment averages an absolute minimum of 3.25 amino acid substitutions per position, thus meeting the criterion of three substitutions per position derived by Greenblatt et al for an alignment that is sufficiently informative to make predictions based on sequence conservation.30 There are 130 invariant positions in the alignment. The con-LR at these invariant positions is 27, indicating odds of 27:1 that the individual invariant positions are functionally constrained. The con-LR drops to 2.6, 0.25, 0.025, and 0.003 at positions where a minimum of 1, 2, 3, and 4 substitutions are required in order to account for the observed alignment, respectively. Thus the likelihood that a position in BRCA1 is under strong functional constraint drops steeply as the number of evolutionarily tolerated substitutions at that position increases.
Grantham deviations of known deleterious, unclassified, and known neutral missense substitutions are displayed in fig 2. As GDs are calculated for successively more informative iterations of the multiple sequence alignment, a dramatic trend in the data becomes apparent: GDs for known deleterious mutations are essentially the same as standard Grantham differences and change very little as further diverged sequences are included in the analysis. In contrast, GDs for known neutral substitutions are lower than standard Grantham differences and drop precipitously as further diverged sequences are included in the analysis. This is explained by visualising the bounding boxes containing positions in the alignment at which deleterious and neutral substitutions have been observed. Because there is very little cross-species sequence variation at the positions at which most deleterious BRCA1 substitutions have been observed,19 the corresponding bounding boxes remain small. In contrast, there tends to be considerable sequence variation at the positions of known neutral substitutions. At these positions, as we consider greater evolutionary breadth, the corresponding bounding boxes expand and in so doing include the space into which many possible missense substitutions fall.
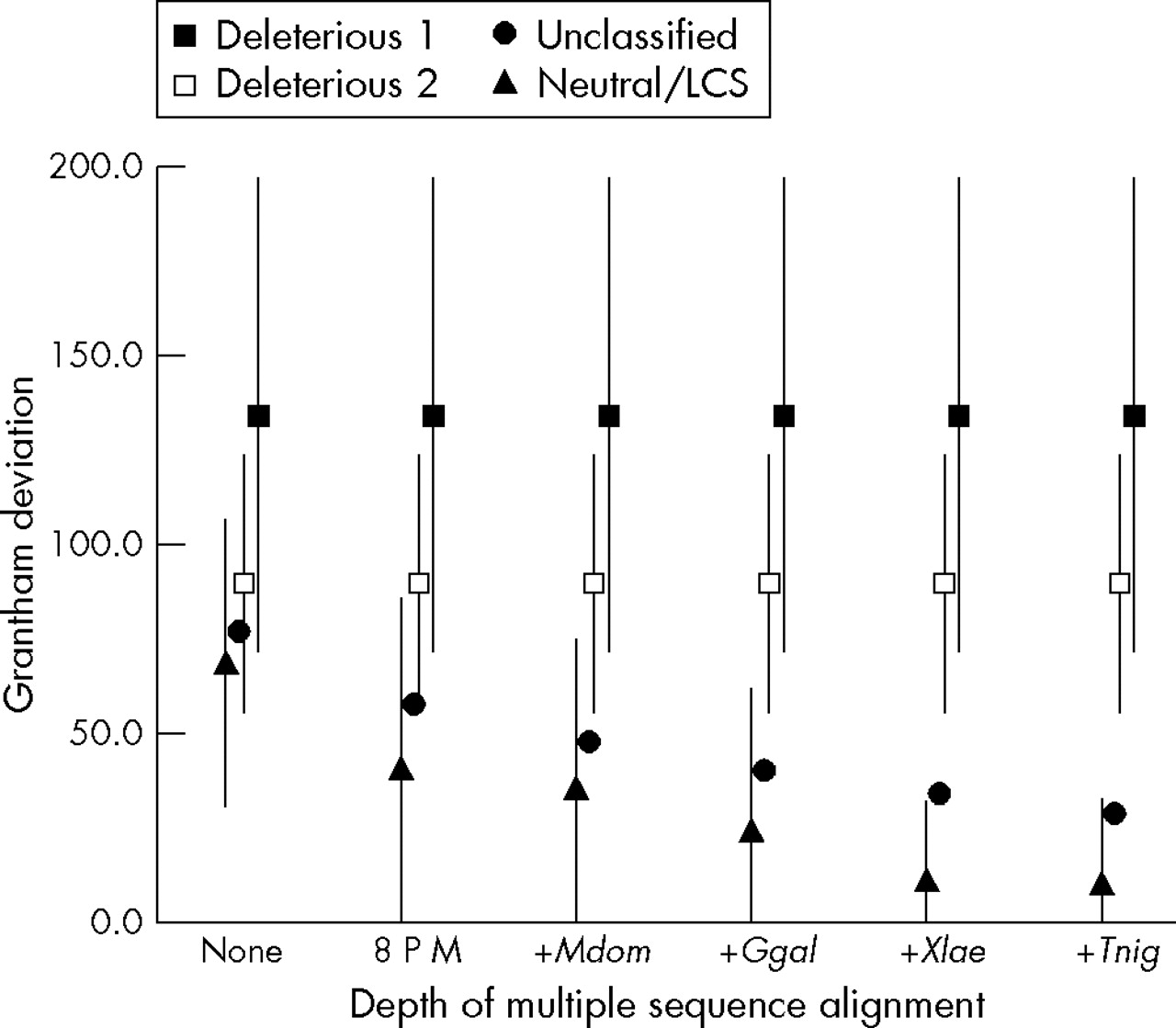
A-GVGD analysis of missense substitutions in BRCA1. Excluding missense substitutions within 2 bp of a splice junction, there were a total of 438 unique BRCA1 missense substitutions in the B1&2 40K set. Using the BRCA1 protein multiple sequence alignment and Grantham deviations (GDs), we have divided these into four data series. The four data series are: filled squares, missense substitutions coded as deleterious, but excluding those that fall within 2 bp of a splice junction; open squares, missense substitutions coded as deleterious, but excluding those at the initiator methionine, those that alter a canonical C3HC4 cysteine residue, and those that fall within 2 bp of a splice junction; closed circles, unclassified missense substitutions, but excluding those that fall within 2 bp of a splice junction; closed triangles, substitutions that are coded as neutral/LCS. In this figure, the x axis represents the depth of alignment at which each analysis was made. None, no alignment (that is, standard Grantham differences); 8 P M, alignment of eight placental mammals; +Mdom, addition of M domestica (opossum); +Ggal, addition of G gallus (chicken); +Xlae, addition of X laevis (frog); +Tnig, addition of T nigroviridis (pufferfish). Error bars give ±1 standard deviation of GD except that some lower error bars are cut off at 0 because GDs cannot take a negative value. Error bars for the unclassified variant data series (closed circles) were omitted because they would tend to obscure evidence that the GDs of known neutral variants are resolved from those of known deleterious variants.
We could, in principle, model the distribution of GD data, estimate the probability density functions for the distributions of scores for known neutral/LCS and deleterious variants, and then format a likelihood expression. However, such a likelihood expression would not be independent of the con-LR because both depend on sequence variation in the alignment. Thus the con-LR and a likelihood expression based on GDs cannot be used as independent factors in an integrated analysis. Simultaneous use requires that they be combined in a way that accounts for this co-dependence, which is beyond the scope of this analysis. We note, however, that evidence in favour of neutrality will be greatest when GD = 0. Hence, if we use the con-LR by itself in these cases it will be a very conservative approximation to the likelihood ratio that the combined analysis would provide. Until a complete likelihood expression has been developed, we will regard the con-LR as inappropriate for the application of identifying neutral variants if the GD>0.
The appropriate evolutionary tree
The last question is whether data from all species in the BRCA1 sequence alignment should be used to calculate GV and GD. Beyond the alignment of eight placental mammal BRCA1s, the average GD for known neutral/LCS variants drops with the addition of each further diverged sequence. The sequential decrease with addition of the opossum, chicken, and frog sequences is substantial and the standard deviations of the GD also decrease. In contrast, the decrease in both GD and its standard deviation with the addition of the pufferfish sequence is quite small (fig 2).
One of the more difficult issues in BRCA1/2 genetics has been estimation of odds ratios for deleterious mutations. The basic problem is that, with a summed allele frequency for all the high risk missense substitutions of <<1%, testing the required number of controls is prohibitively expensive. Because BRACAnalysis is a full sequence test of both BRCA1 and BRCA2, with results tracked in a single database, we know which carriers of interesting sequence variants in BRCA1 also carried a clearly deleterious mutation in BRCA2. Under the hypothesis that the breast/ovarian cancer risk for a BRCA1:BRCA2 double carrier is not dramatically higher than the risk for a simple BRCA2 carrier, the appearance of a double carrier in the B1&2 40K set is largely explained by the deleterious BRCA2 variant. Hence, we can use the BRCA1 chromosomes of the BRCA2 mutation carriers as a kind of control and thereby calculate a BRCA1 with BRCA2 ascertainment-for-BRACAnalysis odds ratio (B1:2 A-OR). That the underlying biological hypothesis is reasonable follows immediately from the observation that the two genes function in the same biochemical pathway and loss of function of the good copy of BRCA1 or BRCA2 is unlikely to be either the initiating or the rate limiting step of tumourigenesis in mutation carriers (for discussion see Venkitaraman31).
Twelve of the known neutral missense substitutions in BRCA1 have allele frequencies of <10%. Many of the carriers of one or another of these neutral BRCA1 substitutions also carry a clearly deleterious mutation in BRCA2. If we use this stratum as the reference category, then the B1:2 A-OR for the four common neutral missense substitutions in BRCA1 is 0.98 (95% CI 0.9 to 1.1). In contrast, the B1:2 A-OR for all truncating mutations in BRCA1 is 9.2 (95% CI 6.4 to 13.2). The B1:2 A-OR is emphatically not the odds ratio for carriage of a variant of interest in BRCA1, but it may well track with the true odds ratio. Figure 3 gives B1:2 A-ORs for missense substitutions at positions in the alignment that are invariant, missense substitutions that are outside the range of variation observed at variable positions in the alignment (that is, GV>0 and GD>0), and missense substitutions that are inside the range of variation observed at variable positions in the alignment (that is, GV>0 and GD = 0). Two points emerge. First, many of the missense substitutions falling at invariant positions in the alignment must be deleterious and the longer the period over which the position has been invariant, the stronger the evidence that this is so. Second, the pooled evidence is in accord with a hypothesis that missense substitutions that fall at variable positions in the alignment of vertebrate BRCA1s and are within the range of variation observed at those positions are neutral. Perhaps surprisingly, even the 36 additional missense substitutions that are brought within the range of variation by the step from frog to pufferfish have a B1:2 A-OR, as a group, of 0.89 (95% CI 0.48 to 1.65). Thus, for the narrow application of validating the con-LR, our results including pufferfish are not substantially different than those that include Xenopus but exclude pufferfish. Nevertheless, we note that the B1:2 A-OR for variable position substitutions that are outside the range of variation at those positions takes a sharp (and possibly important) upswing in the step from Xenopus to pufferfish, suggesting that the validity of the analysis might be reaching some sort of limit at this evolutionary distance. If so, then future data from even more distant (for example, non-vertebrate) genomes might clearly reveal this limit by adding variants that abruptly change the “curves” for the variable sites both within and outside the range of variation. In view of the objective of providing patients and their health-care advisors with unambiguous guidelines for interpreting the implications of rare BRCA1 variants (as neutral versus deleterious), we believe that truncating the analysis of GD between Xenopus and pufferfish makes the best sense, at least until sequence data from more distant species are analysed and utilised in these calculations.

{kind=link}
{kind=link}
{kind=link}
Ascertainment risk associated with groups of missense substitutions in BRCA1. Excluding missense substitutions at the initiator methionine, within 2 bp of a splice junction, and those already classified as neutral/LCS, there were a total of 418 unique BRCA1 missense substitutions in the B1&2 40K set. Using the BRCA1 protein multiple sequence alignment and A-GVGD, we have divided these into three data series. The three data series are: closed triangles, substitutions that fall at variable positions in the alignment and are within the observed range of variation to that layer in the alignment (GV>0, GD = 0); closed circles, substitutions that fall at variable positions in the alignment and are outside the observed range of variation to that layer in the alignment (GV>0, GD>0); and closed squares, substitutions that fall at positions that are invariant to that layer in the alignment (GV = 0, GD>0). In this figure, the x axis represents the depth of alignment at which each analysis was made. 8 P M = alignment of eight placental mammals; +Mdom, addition of M domestica; +Ggal, addition of G gallus; +Xlae, addition of −X laevis; +Tnig, addition of T nigroviridis. The reference set (B1:2 A-OR = 1.0) was the group of 12 known neutral missense substitutions in BRCA1 that have allele frequencies of <10%. Error bars on the closed circle and closed triangle data series are the 95% confidence interval adjusted for a three degree of freedom two-sided test; error bars for the unclassified variant data series (closed circles) were omitted because they would tend to obscure evidence that the B1:2 A-ORs of known neutral variants are resolved from those of known deleterious variants.
Grantham analysis
Of the 15 neutral/LCS missense substitutions that we analysed for co-occurrence, 14 fall at positions in the protein that have substantial cross-species sequence variability. Twelve of these have a con-LR of ⩽0.003 and the other two have con-LR = 0.025. The remaining neutral variant, P871L, falls at a position that is leucine in all the other species in the alignment. Eleven of these 15 neutral/LCS missense substitutions, including P871L, have GDs of 0 in the alignment from human to Xenopus. For these 11 substitutions, we conclude that the con-LR is appropriate for inclusion in an integrated analysis (table 3).
Alignment/Grantham analysis of missense substitutions in BRCA1 that have been observed with two or more different known deleterious mutations
Of the 18 unclassified variants that have co-occurred at least twice with a clearly deleterious BRCA1 mutation, 14 fall at positions in the protein that have substantial cross-species sequence variability. Seven have a con-LR of ⩽0.003 and seven have con-LR = 0.025. Nine of the 14 missense substitutions with relatively low con-LRs also have GDs of 0 in the alignment from human to Xenopus. For these nine substitutions, we again conclude that the con-LR is appropriate for inclusion in an integrated analysis (table 3).
Integrated analysis
Integration of the co-occurrence data with the alignment/Grantham data is achieved by simply multiplying the two likelihood ratios, taking into account whether the con-LR was appropriate. Goldgar et al7 discussed and then set thresholds for declaring an unclassified variant either deleterious or neutral/LCS on the basis of the analysis. If the integrated likelihood is >1000 or <0.01, then the variant is considered deleterious or neutral/LCS, respectively. If the score is between those two thresholds, the variant remains unclassified. In the present two method analysis, when the con-LR is inappropriate, we consider that only one valid analysis has been done. Similarly, if the observed co-occurrences did not meet our precautionary criteria, we consider that only one valid analysis has been done. As neither of these two cases are an integrated analysis, the variant would remain unclassified.
Of the 15 known neutral/LCS variants that we analysed, 11 would have been classified as neutral/LCS by the approach taken here (table 4). The four substitutions that would not have been classified neutral/LCS fail because the human missense substitution is outside the range of variation observed from human to frog.
Integrated analysis
For eight of the 18 unclassified variants that we analysed, we calculated valid likelihood ratios below 0.01. We conclude that these missense substitutions are neutral/LCS (table 4). The B1:2 A-OR for these eight substitutions is 0.89 (95% CI 0.55 to 1.44) also indicating that, in aggregate, this group of variants confers no greater risk of familial breast/ovarian cancer than do the other missense substitutions that have already been classified neutral/LCS. The human missense substitutions defining nine of the other unclassified variants fall outside the range of variation observed from human to frog; these remain unclassified. One additional substitution, R504H, met the criterion of GD = 0 but remains unclassified because it is almost certainly in cis with the deleterious variant with which it has repeatedly co-occurred. The B1:2 A-OR for this group of 10 substitutions is 1.15 (95% CI 0.54 to 2.48).
The eight substitutions that we have classified as neutral/LCS were observed a total of 346 times in 40 000 tests, 268 times with no clearly deleterious mutation in BRCA1 or BRCA2. However, fewer than 268 unclassified variant reports are resolved because some patients carried more than one unclassified variant. For example, Y179C, F486L, and N550H are often seen together and probably constitute a rare haplotype. One of these three substitutions, F486L, met the criteria for neutral/LCS, but the other two did not. Thus the unclassified variant reports on the 38 individuals who carried all three of these substitutions, but no clearly deleterious variant, are not resolved. In the end, we find 202 patient reports that would move from containing a reportable unclassified variant to no reportable variants as a consequence of this analysis.
DISCUSSION
We conclude that eight currently unclassified missense substitutions in BRCA1 (L246V, F486L, R496H, M1008V, M1008I, E1250K, D1546N, and L1564P) should be considered neutral/LCS. Our approach to classifying these missense substitutions might seem unusual to some because we used neither segregation analysis nor association study nor functional assay. Instead, we used two methods, co-occurrence with clearly deleterious mutations in BRCA1 and A-GVGD. The analysis was carried out under the assumption that neither method is inerrant.
Co-occurrence is really a test for embryonic lethality due to inheritance of a compound heterozygous null genotype. The underlying assumption is that inheritance of a genuine high risk missense substitution in BRCA1, along with a clearly deleterious mutation, will either lead to death during embryogenesis or a severe phenotype such as Fanconi’s anaemia. Internal statistics of the BRACAnalysis database provide support for this hypothesis. But it is difficult to exclude the possibility that some BRCA1 genotype combinations of interest could result in a high cancer risk with no other obvious phenotype. The method also has the pitfall that the clearly deleterious mutation and the sequence variant of interest must be in trans for the analysis to be valid. On this latter point we have taken the cautious approach of only analysing missense substitutions for which at least two co-occurrences have been observed and, when the number of distinct co-occurrences was very small, confirming by SNP haplotype analysis that at least one was in trans.
In order to measure the range of sequence variation that has occurred at specific residues in BRCA1 during vertebrate evolution and the fit between observed missense substitutions and that range of variation, we extended the concept of Grantham scores to multiple sequence alignments. The method, A-GVGD, can be used to identify sets of missense substitutions that are either enriched for deleterious variants or enriched for neutral variants. However, A-GVGD does not account for the possibility that sequence variation that has been permissible during the evolution of BRCA1 in one group of non-human vertebrates is not permissible in human BRCA1. It also does not take into account that the nucleotide substitution underlying a missense variant may interfere with mRNA splicing or have some other deleterious effect at the level of DNA or RNA.
In integrating these methods, we required that the cooc-LR is <1 and that GD = 0. Thus we only make the classification in those eight cases where combined evidence from co-occurrence data, variability in the sequence alignment, and GD, support the conclusion neutral/LCS. Essentially, we have used these methods in such a way that each complements the potential for error in the other. The integrated likelihoods for each of these eight missense substitutions are all more than two orders of magnitude below the threshold of 0.01 set by Goldgar et al,7 thus our confidence in the classification, in each of these cases, is very strong. There were 10 unclassified substitutions that we analysed closely and did not classify as neutral/LCS. The B1:2 A-OR for this set of substitutions (1.15) was not much higher than that for the group that we did classify (0.89). There is no substantial evidence that there is more risk associated with the 10 substitutions that we did not classify neutral/LCS than the ones that we did; rather, that these 10 variants remain unclassified may be viewed as an example of how applying very strict criteria in order to minimise type I error can reduce our power to classify substitutions that may well be neutral.
In the 40 000 test data set there remain 39 unclassified missense substitutions that have been observed to co-occur once with a clearly deleterious variant. Nineteen of these have GV = 0 in the alignment from human to Xenopus; consequently, with the pair of methods used here, we would expect to be able to reclassify many of these as neutral/LCS. However, there also remain 164 unclassified missense substitutions with GD = 0. As the B1:2 A-OR for this pool of substitutions is 0.91 (95% CI 0.60 to 1.39), the vast majority of these must be neutral/LCS. That only 19 of these co-occurred with a clearly deleterious mutation underscores the point that further clinical reclassification of superficially innocuous BRCA1 missense variants will require integrated application of other methods such as pooled family histories, segregation analysis, or validated functional assays. At this point, however, BRCA2 is creating a noticeably larger burden of unclassified missense substitutions than BRCA1. Thanks to model organism genome sequencing, sufficient genome sequences are available to allow the straightforward gene model creation, testing, and correction required to compile a BRCA2 protein multiple sequence alignment comparable to the BRCA1 alignment used here. As the same analysis model appears to apply to BRCA2, this should lead to reclassification of a considerable number of missense substitutions followed by a substantial reduction in the burden of tests that include a reportable unclassified variant.
REFERENCES
Footnotes
-
Published Online First 13 July 2005
-
This research was supported in part by the INHERIT BRCAs program from the Canadian Institute for Health Research, grant 7792 (to SVT) from the Association pour la Recherche sur le Cancer, and grant RR14214 (to PBS) from the US National Institutes of Health. None of the funding agencies had any influence on the direction of this work.
-
Competing interests: AMD, TJ, TS, and AZ are employees of Myriad Genetics, Inc., or Myriad Genetic Laboratories, Inc. SVT and AT own stock in Myriad Genetics, Inc. Results from this work bear on Myriad Genetic Laboratories’ commercial test for mutations in BRCA1 and BRCA2.