Article Text

Abstract
Background: The acronym CHARGE refers to a non-random cluster of malformations including coloboma, heart malformation, choanal atresia, retardation of growth and/or development, genital anomalies, and ear anomalies. This set of multiple congenital anomalies is frequent, despite rare patients with normal intelligence, and prognosis remains poor. Recently, CHD7 gene mutations have been identified in CHARGE patients; however, the function of CHD7 during development remains unknown.
Methods: We studied a series of 10 antenatal cases in whom the diagnosis of CHARGE syndrome was suspected, considering that a careful pathological description would shed light on the CHD7 function during development. CHD7 sequence analysis and in situ hybridisation were employed.
Results: The diagnosis of CHARGE syndrome was confirmed in all 10 fetuses by the identification of a CHD7 heterozygous truncating mutation. Interestingly, arhinencephaly and semi-circular canal agenesis were two constant features which are not included in formal diagnostic criteria so far. In situ hybridisation analysis of the CHD7 gene during early human development emphasised the role of CHD7 in the development of the central nervous system, internal ear, and neural crest of pharyngeal arches, and more generally showed a good correlation between specific CHD7 expression pattern and the developmental anomalies observed in CHARGE syndrome.
Conclusions: These results allowed us to further refine the phenotypic spectrum of developmental anomalies resulting from CHD7 dysfunction.
- CHARGE syndrome
- CHD7 gene
- development
- gene expression pattern
- human embryogenesis
Statistics from Altmetric.com
CHARGE syndrome refers to an association of defects including ocular coloboma (C), heart disease (H), choanal atresia (A), retarded growth and/or anomalies of the central nervous system (R), genito-urinary defects and/or hypogonadism (G), and ear anomalies and/or deafness (E). The acronym was coined by Pagon et al,1 but the syndrome was first reported by Hall2 and Hittner et al.3 Other diagnostic criteria were proposed based on major/minor anomalies,4–6 however, all features have variable expression and can be inconsistent, and many are non-specific. The incidence of CHARGE syndrome has been estimated to range from 0.1 to 1.2/100 000 live births with the highest incidence (to 1/8500 live births) in Canada.7
Recently, Vissers et al reported mutations in the CHD7 gene in two out of three CHARGE patients tested.8 CHD7 belongs to a large family of evolutionarily conserved proteins thought to play a role in chromatin organisation. However, although more than 200 newborns and infants with CHARGE syndrome have been described, only two prenatally detected cases have been reported to date9,10 and nothing is known about CHD7 function during fetal development. In the present study, we analysed the coding sequence of the CHD7 gene in 10 fetuses presenting with clinical features of CHARGE syndrome, considering that a careful pathological description would shed light on the CHD7 function during development. Clinical features as well as criteria necessary to establish the diagnosis of CHARGE syndrome prenatally remained to be defined. In fetuses, coloboma, heart malformation, choanal atresia, intrauterine growth retardation, genital anomalies, and external ear anomalies can be identified, while clinical symptoms such as mental retardation and deafness have to be replaced by their equivalents: central nervous system (CNS) anomalies and semi-circular canal (SCC) hypoplasia or agenesis, respectively. In this series, four of the above major anomalies were required for the diagnosis of CHARGE syndrome because the other features, such as growth retardation, cryptorchidism, or cranial nerve dysfunction, appear only in the postnatal period. All fetuses underwent autopsy and a detailed neuropathological examination with close attention to olfactory bulbs and tracts, brainstem, and cerebellum.
We identified a truncating mutation in all cases examined, confirming the diagnosis of CHARGE syndrome in all 10 fetal cases and allowing the phenotypic spectrum of the developmental anomalies resulting from CHD7 dysfunction to be further envisaged. In addition, in situ hybridisation analysis of the CHD7 gene at different stages of early human development emphasised the role of CHD7 in the development of the central nervous system, internal ear, and neural crest of pharyngeal arches, and more generally showed a good correlation between specific CHD7 expression pattern and the developmental anomalies observed in CHARGE syndrome.
METHODS
Patients
In all cases, the presence of severe malformations was noted at ultrasound (US) examination and pregnancies were terminated in accordance with French law. Detailed clinicopathological examination was carried out after parental consent. For all cases, the diagnosis of CHARGE syndrome was established either based on US evaluation or after fetopathological examination using the diagnostic criteria described above. For all cases, karyotype and CHD7 mutation screening was performed. Clinical and genetic data are summarised in table 1 and detailed in supplementary data available online at http://www.jmedgenet.com/supplemental.
Summary of the clinical features of the 10 CHARGE fetuses
CHD7 sequence analysis
DNA was extracted from frozen tissues according to standard protocols. We designed 42 primer pairs covering the 37 CHD7 coding exons (exons 2–38) and the corresponding exon-intron boundaries. Primer sequences and PCR conditions are available on request. PCR products were purified and directly sequenced in both directions on an ABI PRISM 3730 DNA sequencer (Perkin Elmer-Applied Biosystems, Courtaboeuf, France) using the dye terminator method according to the manufacturer’s instructions.
In situ hybridisation
Normal human embryos and fetal tissues were obtained after elective termination of pregnancy in agreement with French legislation (law no. 94-654 of July 29, 1994), Necker Hospital, and the National Ethics Committee recommendations (report no. 1 of May 22, 1984). Embryonic stages were established according to the Carnegie staging (CS) classification.11 Seven different embryonic stages (CS9 (d20), CS10 (d22), CS11 (d24), CS12 (d26), CS14 (d33), CS15 (d34), and CS19 (d 47–48)) as well as two fetal stages (9 and 11 weeks) were studied. Tissues were fixed in 4% phosphate buffered paraformaldehyde, dehydrated, and embedded in paraffin blocks. Five micron thick serial sections were cut. Exon 35 primers were selected for PCR amplification (F: 5′-GCTGTTCCCAAACAACTAGACATTG-3′ R: 5′-GAAACATTCAAGGAAAAGGCAGAG-3′). A T7 promotor sequence extension (TAATACGACTCACTATAGGGAGA) was added at the 5′ end of each primer. T7F/R and F/T7R primers allowed the amplification of sense and antisense templates specific to the CHD7 gene. Riboprobes labelling, tissue fixation, hybridisation, and developing were carried out according to standard protocols,12 as previously described.13
RESULTS
Molecular results
Direct sequencing of the 37 CHD7 coding exons (gene ID 55636, NM_017780, NCBI: http://www.ncbi.nlm.nih.gov/) detected ten heterozygous truncating mutations. They are summarised in fig 1. Six were nonsense mutations (cases 2, 3, 4, 7, 8, 10), and four were frameshift mutations predicting a premature stop codon (cases 1, 5, 6, 9). In seven cases, parental DNA was studied (cases 1, 3, 6, 7, 8, 9, 10). All mutations occurred de novo (table 1). None of the 10 mutations was described previously. We found two mutations in exon 2, two mutations in exon 8 (at the end of the first chromodomain), and two identical mutations in exon 12 (R987X, unrelated cases 2 and 8) located in the SNF2 domain. In patient 5, the mutation (N1371fsX1374) is located in another domain of CHD7 encoding the putative helicase domain. None of the remaining mutations involved known functional domains of the protein.
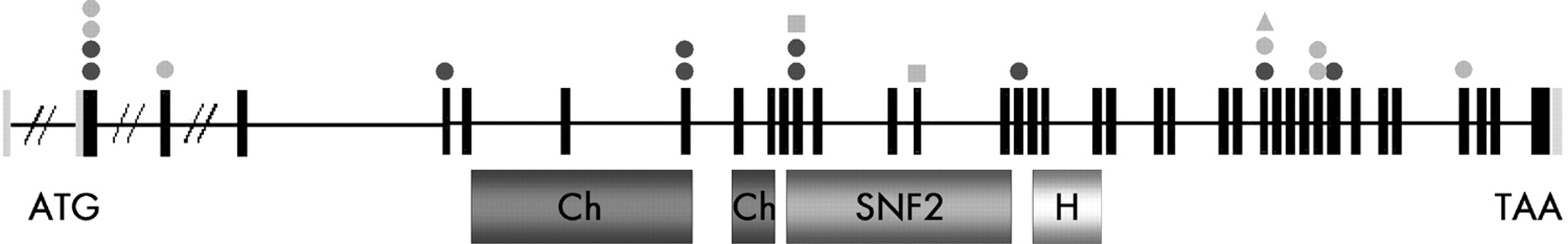
Schematic representation of the CHD7 mutations identified so far. The mutations found by Vissers et al8 are shown in orange; the ten nonsense mutations found in our cohort are shown in blue. Circles indicate the nonsense mutations, squares the missense mutations, and triangles the intron-exon boundary mutations. On the bottom, functional CHD7 domains are indicated: chromodomains in red, SNF2 domain in green, and helicase domain in yellow.
Clinical analysis
This series of 10 fetuses (seven male and three female) ranged from 21 to 36 weeks of gestation (WG) with a mean of 27 WG. Mean paternal age was 35 years and 8 months and mean maternal age was 30 years and 6 months. Prenatal US was abnormal in all patients. It disclosed polyhydramnios in 3/8 cases. Although two fetuses (cases 7 and 9) weighed between the 3rd and the 10th percentile, intrauterine growth retardation was never observed. All fetuses presented bilateral and asymmetric external ear abnormalities, semi-circular canal hypoplasia/agenesis, and arhinencephaly. As shown in fig 2A, the ears were typically low set, asymmetric with a small or absent lobe, posteriorly rotated and triangular or square shaped.
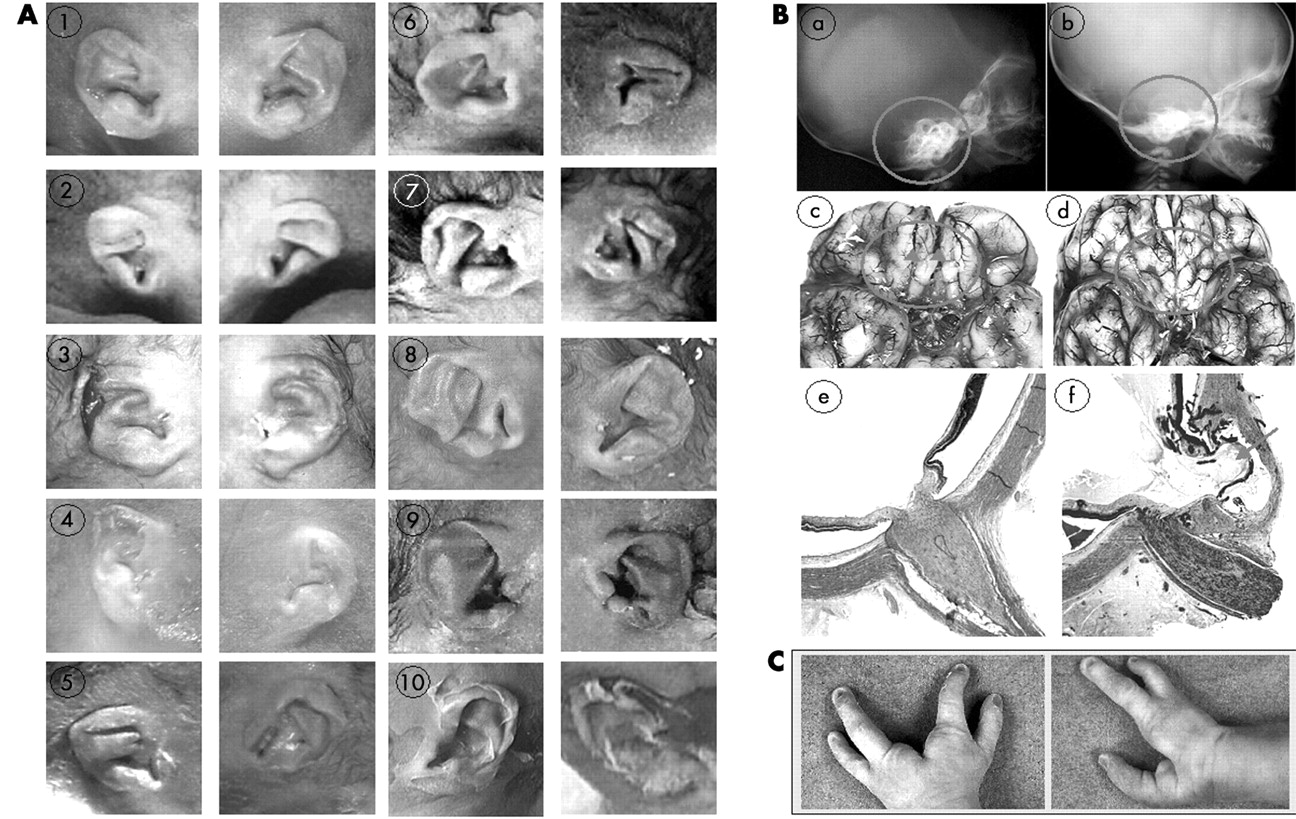
(A) External ear dysmorphism in the 10 fetuses. Note the characteristic triangular or square shaped and wrinkled morphology in all cases. (B) Normal (on the left) and pathological (on the right) view of three of the major clinical findings, namely severe semicircular canal hypoplasia (b: fetus 10) on profile babygram at 19 weeks (control at 19 weeks), arhinencephaly (d: fetus 10) at 34 weeks (control at 34 weeks), and retinal coloboma (f: fetus 1) at 19 weeks (control at 19 weeks). (C) Hands of fetus 9 showing the ectrodactyly. (These images are published with consent.)
Coloboma was found in seven patients, always affecting the retina or the choroid segment (fig 2Be,f). Microphthalmia was observed in two out the 10 cases in accordance with earlier postnatal observations.14
A congenital heart defect was found in 9/10 patients. These defects were complex cardiopathies and five involved the conotruncal region (cases 2, 3, 4, 5, 9). They frequently included atrial and/or ventricular septal defects, in accordance with earlier postnatal observations.15
Choanal atresia was found in six out of 10 patients (four bilateral and two unilateral). Among the four remaining patients, three had a cleft lip and/or palate. In two cases unilateral choanal atresia was found with a controlateral cleft palate. In one patient (case 6), neither choanal atresia nor facial clefting was found.
CHD7 expression during human embryonic development
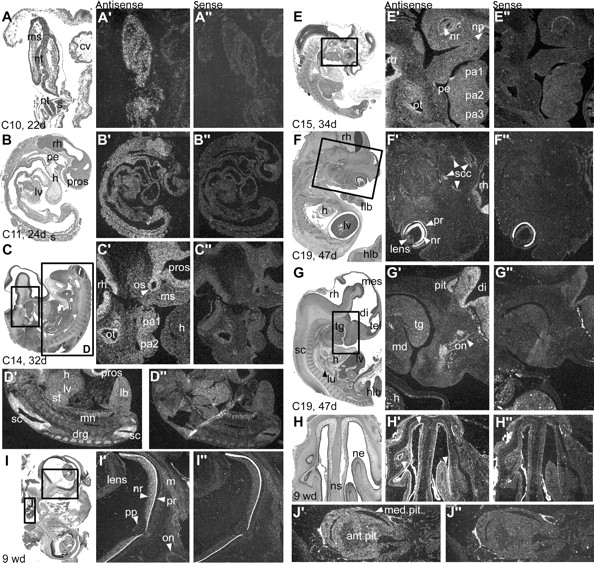
At CS9 (d20; not shown), CS10 (d22; fig 3A), and CS11 (d24; fig 3B), CHD7 is ubiquitously expressed, with a distinct signal in the neural tube. At CS12 (d26), CHD7 is expressed throughout the CNS and neural crest-containing mesenchyme of the pharyngeal arches. At CS14 (d33), CHD7 transcripts continue to be expressed in the cephalic mesenchyme, pharyngeal arches, and the brain. Expression is observed in the otic vesicle (fig 3C) as well as in the limb bud mesenchyme; it is more intense in the spinal cord and dorsal root ganglia (fig 3D). By CS15 (d34; fig 3E), expression continues to be intense within the CNS but is restricted to the dorsal part of the otic vesicle. Interestingly, no expression is observed in the nasal placode, neural retina, or pharyngeal endoderm. Also no expression is detected in the heart at any age. By CS19 (d47), CHD7 is strongly expressed in the neural retina and rhombencephalon. It is moderately expressed in the semicircular canals (fig 3F), as well as in the forebrain, pituitary gland, and olfactory bulbs and nerves (fig 3G). Expression at 9 weeks of development is localised in the nasal epithelia (fig 3H), the neural retina, the optic nerve sheath (fig 3I), and the anterior and median lobes of the pituitary gland (fig 3J).

{kind=link}
{kind=link}
{kind=link}
Expression of the CHD7 gene during human embryonic development. Adjacent sections are coloured with haematoxylin/eosin (X), treated with antisense or sense control riboprobes (X′ and X′′, respectively). Ventral to right, rostral to top where applicable. (A) At Carnegie stage (C)10, after 22 days (d) of development, CHD7 is expressed ubiquitously within embryonic but not extraembryonic tissues (cv) seen in transverse section. (B) By C11, expression is slightly more intense within the rhombencephalon, as observed in parasagittal section, but is not markedly present within the heart (h), liver (lv), or pharyngeal endoderm (pe). (C) A stronger signal is observed in the entire central nervous system (prosencephalon (pros) and rhombencephalon (rh)) by C14, including the diencephalic optic stalk (os), and is also seen in the cephalic mesenchyme (ms) of the frontonasal bud and pharyngeal arches (pa1, pa2). Heart expression is non-existent/not found (h). The otic vesicle, negative at C12 (not shown), expresses CHD7 in its epithelium (ot) and associated acoustic ganglion (*) at this stage. Parasagittal section. (D) Enlargement of the body of the same C14 embryo shows a strong CHD7 signal in the spinal cord (sc) and dorsal root ganglia (drg), and a weak signal in limb bud mesenchyme (lb). (E) At C15, CHD7 expression is restricted to the dorsal epithelium of the otic vesicle (ot), presumptive inner ear, and the rhombencephalon (rh), spinal cord, and dorsal root ganglia (not shown); however, expression is barely above background in pharyngeal arch mesenchyme (pa1–3) and absent from the neural retina (nr), pharyngeal endoderm (pe), and nasal placode (np). (F, G) By C19, the CHD7 signal becomes intense in the entire neural retina (nr), rhombencephalon (rh), anterior pituitary gland (pit), diencephalon (di; in particular the hypothalamus (hyp)), and olfactory nerve (on, arrowhead) and bulb and is more discrete but distinct within the semicircular canals (scc). Expression continues to be absent from any portion of the heart (h). md, mandible; tg, tongue. (H) At 9 weeks of development (wd), CHD7 transcripts are detectable in the nasal epithelium (ne) but not within the nasal septum (ns) in coronal section. (I) Also at 9 wd, CHD7 expression is observed in neuronal soma of the neural retina in contrast to the absence of signal in the axon fascicles at the optic papilla (pp) and nerve (on), although the outer sheath of the nerve continues to express CHD7 (compare frame C′). (J) An enlargement of the pituitary gland in the same coronal section as (I) demonstrates continued CHD7 expression within anterior and median lobes at 9 wd.
DISCUSSION
Here we report on a series of fetuses with CHARGE syndrome using diagnostic criteria adapted to the prenatal situation. All fetuses underwent an extensive necropsy as well as molecular analysis of the CHD7 gene. This study confirms the role of the CHD7 gene in CHARGE syndrome and allows delineation of the antenatal clinical spectrum of CHD7 mutations.
Molecular analysis of CHD7
Nine different mutations were identified in our series. One, the R987X mutation, was found in two unrelated fetuses (cases 2 and 8). Interestingly, they differed as regards the presence of distal arthrogryposis and hemivertebra (case 2), suggesting that other additional factors might be involved. However, based on our results and in agreement with Vissers et al, so far it is not possible to establish a genotype/phenotype correlation.8 Large deletions and nonsense mutations in the first exon(s) of the CHD7 gene are observed. This suggests that haploinsufficiency of the CHD7 gene is probably the mechanism accounting for CHARGE syndrome. Because the mutations occurred de novo, genetic counselling can be reassuring, although germ-line mosaicism can never be excluded.
Clinical analysis
In our series of fetal CHARGE syndrome, our detailed clinicopathological evaluation led to the identification of three constant features: anomalies of the external ear, agenesis/hypoplasia of the semi-circular canals, and arhinencephaly. Six other features occur frequently (in at least in seven of 10 cases), namely genital anomalies, thymic hypoplasia, ocular coloboma, other CNS anomalies (other than arhinencephaly), choanal atresia/cleft palate, and heart defect.
Bilateral and asymmetric external ear malformations are found in all our cases. As shown in fig 2A, typical features are small, low set, posteriorly rotated ears, with a prominent crux helix, a small or absent lobe, and a triangular shape as described previously.5,15,16 In the literature, internal ear anomalies, ranging from subtle modular deficiencies to Mondini malformation and semi-circular canal hypoplasia or agenesis, are classically reported in CHARGE syndrome.7,17,18 Semi-circular hypoplasia/agenesis was reported in up to 100% of cases in the literature.15,18 We confirm these data and emphasise the necessity of detecting temporal bone anomalies though cephalic x ray evaluation for the antenatal diagnosis of CHARGE syndrome (fig 2Ba,b).
Interestingly, among CNS anomalies, arhinencephaly was found in all our cases (fig 2Bc,d). Arhinencephaly has already been reported by Lin et al in some patients.19 However, its frequency among CHARGE patients has not been evaluated in the literature, aside from the observation of Harvey et al who found arhinencephaly in 7/7 CHARGE postnatal cases.20 More recently, Chalouhi et al assessed olfactory deficiency in 14 children with CHARGE syndrome. Half of them were anosmic and the others had olfactory residual function (hyposmic). All nine MRIs showed anomalies of the olfactory tracts and bulbs varying from moderate hypoplasia to complete aplasia, without any relationship between the radiological and functional results.21 Our systematic neuropathological examination clearly shows that arhinencephaly is a constant sign of CHD7 mutation. We propose it should be considered a major sign for prenatal CHARGE diagnosis. Other CNS anomalies included brainstem and cerebellum abnormalities. They were observed in eight of our 10 fetuses, but were reported also in postnatal cohorts.19,20 They are mainly characterised by hypoplasia of the inferior cerebellar vermis and brain stem (cases 1, 5, 7) and severe cerebellar heterotopia (case 8).
The high incidence of heart defects, cleft lip/palate, and brain anomalies in our series when compared to postnatal data can probably be explained by detection by US during pregnancy. It has been shown that CHARGE individuals have increased mortality due to AVSD defects and cerebellar and or brain stem anomalies associated with ventriculomegaly.7,15 Moreover, males have been reported to have an increased mortality compared to females.15,22 This study presents a population of 10 fetuses with a severe phenotype of CHARGE syndrome, with an especially high number of complex cardiopathies, bilateral posterior choanal atresia, tracheo-oesophageal fistula,22 brain anomalies, and increased representation of males (7/10).
Among other frequent features, genital anomalies deserve special attention: they were found in 7/10 cases. In three male fetuses, they were noticed only at the histological level (Leydig cell rarefaction). To our knowledge these histological anomalies have not been described previously. This may explain the delay in puberty noticed more in males than females that has been reported postnatally.23 In a female fetus, the only genital anomaly found was a hypoplastic ovary. According to the postnatal literature, female genital hypoplasia is rare and micropenis or cryptorchidism are more frequent features observed in males.24
Interestingly, thymus hypoplasia or agenesis was found in seven of out 10 cases. This contrasts with postnatal cases where thymic hypoplasia is rarely reported.5 The association of CHARGE and DiGeorge syndromes reported previously25 suggests a neural crest defect causes the clinical overlap of both syndromes. Whether the CHD7 gene could be responsible for a CHARGE-DiGeorge association should be tested further, particularly in postnatal patients.
Our study also confirms that growth retardation is usually observed postnatally.26 Indeed, no intrauterine growth retardation was observed in our antenatal series. In addition to the severe feeding problems related to gastro-oesophageal reflux and brain stem anomalies, the postnatal growth retardation could be explained in part by a pituitary gland dysfunction. Interestingly, CHD7 gene expression is observed within this tissue during the embryonic period.
It is worth stressing that case 9 presented with ectrodactyly (fig 2C), which has not been reported so far in CHARGE syndrome. Four patients presented limb anomalies although minor, such as clinodactyly. Skeletal (including costal and vertebral defects) or renal anomalies were found in four out of 10 cases. Finally, among other features, tracheo-oesophageal fistulas were noticed twice.
Based on our clinicopathological observations, we consider semicircular canal agenesis as well as arhinencephaly highly predictive diagnostic criteria of CHARGE syndrome. We suggest that they should be added to the two major diagnostic criteria described by Pagon, along with the external ears malformations. Indeed, four of six major criteria of CHARGE (coloboma, choanal atresia and/or cleft lip/palate, heart defect, arhinencephaly, semi-circular canal agenesis, and external ear anomalies) were necessary and sufficient for the diagnosis of CHARGE in our series. As previously suggested for postnatal cases, minor diagnostic criteria such as facial dysmorphism and renal, digestive, and skeletal anomalies should also be considered for fetal CHARGE syndrome in addition to thymus hypoplasia/agenesis and polyhydramnios. Interestingly, in most patients with three major features and three minor features proposed as CHARGE syndrome,5 the presence of arhinencephaly and semi-circular canals hypoplasia/agenesis was not evaluated and we recommend brain MRI to assess the diagnosis of CHARGE syndrome in these cases.
Our study contributes to the development of a strategy for the diagnosis of CHARGE syndrome during pregnancy. Indeed, features of CHARGE syndrome detected at routine US such as hydramnios, heart defects, cleft lip/palate, CNS anomalies, and kidney or gastrointestinal anomalies are common. More specific features would help in the diagnosis of CHARGE syndrome. We thus propose that focussed US and/or brain MRI should be performed for the detection of external ear anomalies, choanal atresia, semi-circular canal agenesis, and arhinencephaly. This strategy will certainly lead to a higher prenatal detection rate of CHARGE syndrome.
Embryonic function of CHD7
The CHD7 protein encompasses several important domains. The chromatin organisation modifier (chromo) domain is a conserved region of around 50 amino acids found in a variety of chromosomal proteins that appear to play a role in the functional organisation of the eukaryotic nucleus. Experimental evidence shows that the chromodomain is involved in binding proteins to histone and possibly RNA. CHD7-like helicase domains are involved in ATP-dependent unwinding of DNA or RNA duplexes and histone deacetylation.27,28 Certain CHD proteins have been shown to participate in nucleosome remodeling deacetylase (NuRD) protein complexes which interact with sequence-specific DNA-binding factors for targeted repression.29 Presumably, the CHD7 protein plays an important role in chromatin remodelling during early development and allows a level of epigenetic control over target genes expressed in mesenchymal cells derived from the cephalic neural crest.
We analysed the expression pattern of the CHD7 gene during early human development. CHD7 is widely expressed in the undifferentiated neuroepithelium and in mesenchyme of neural crest origin. Towards the end of the first trimester it is expressed in dorsal root ganglia, cranial nerves/ganglia, and auditory, pituitary, and nasal tissues as well as in the neural retina. Absent from the myocardium, bones of mesodermal or neural crest origin, and the genital ridge, CHD7 expression correlates with defects observed in these tissues because of its presence in neural crest cells investing the outflow tract of the heart, and in the hypothalamus and pituitary gland. Endocrine deficiency may occur centrally, in the differentiation of hypothalamic nuclei secreting somatostatin or GnRH, or more peripherally in the differentiation of somatotropic or gonadotropic cells of the anterior pituitary. It could be related to the clinical findings of delayed puberty in the adolescent and adult population with CHARGE syndrome.23 A major testable hypothesis in this context is that CHD7 regulates paired domain- or homeobox domain-containing transcription factor genes important for the development of the pituitary gland and other organ systems, such as Hesx1, Otx1, Prop1, Krox20, Pitx2, and Titf1/Nkx2a. Other regulatory transcription factor genes such as Pax2 or Tbx1, with multiple expression sites in many affected organ systems or inductive tissues for these organ anlages, still remain attractive and non-exclusive CHD7 functional targets.30
CONCLUSION
In the present study we confirmed the predominant role of the CHD7 gene during fetal development. The clinicopathological spectrum of CHD7 mutations in a series of 10 fetuses examined at the anatomical as well as histological level allowed the phenotypic spectrum of the developmental anomalies resulting from CHD7 dysfunction to be further envisaged and led us to propose agenesis of the semi-circular canals and arhinencephaly as major diagnostic criteria of CHARGE syndrome. Further postnatal studies are needed to confirm these data.
Acknowledgments
We are thankful to Eric Detrait, Sophie Delahaie, Boris Keren, Daniel Sidi, the French patients’ group for the CHARGE association, and the SOFFOET (Société Française de FOETopathologie) for their help and collaboration. We also thank Han Brunner for sharing unpublished data.
REFERENCES
Supplementary materials
Files in this Data Supplement:
- view PDF - Cases 1-10.
Footnotes
-
↵* These authors contributed equally to this work
-
Published Online First 23 September 2005
-
DS was supported by the Programme de Recherche Clinique AOM 02-122. HCE was supported by the Association Française contre les Myopathies and the INSERM Avenir program
-
Competing interests: none declared
-
Consent has been given for the publication of the details in this report