Article Text

Abstract
Background: Fryns syndrome (FS) is the commonest autosomal recessive syndrome in which congenital diaphragmatic hernia (CDH) is a cardinal feature. It has been estimated that 10% of patients with CDH have FS. The autosomal recessive inheritance in FS contrasts with the sporadic inheritance for the majority of patients with CDH and renders the correct diagnosis critical for accurate genetic counselling. The cause of FS is unknown.
Methods: We have used array comparative genomic hybridisation (array CGH) to screen patients who have CDH and additional phenotypic anomalies consistent with FS for cryptic chromosome aberrations.
Results: We present three probands who were previously diagnosed with FS who had submicroscopic chromosome deletions detected by array CGH after normal karyotyping with G-banded chromosome analysis. Two female infants were found to have microdeletions involving chromosome band 15q26.2 and one male had a deletion of chromosome band 8p23.1.
Conclusions: We conclude that phenotypes similar to FS can be caused by submicroscopic chromosome deletions and that high resolution karyotyping, including array CGH if possible, should be performed prior to the diagnosis of FS to provide an accurate recurrence risk in patients with CDH and physical anomalies consistent with FS.
- array CGH, array comparative genomic hybridisation
- BAC, bacterial artificial chromosome
- CCD, charge-cooled device
- CDH, congenital diaphragmatic hernia
- CHR, Committee for Human Subjects Research
- CRL, crown-rump length
- FISH, fluorescence in situ hybridisation
- FS, Fryns syndrome
- PCR, polymerase chain reaction
- array comparative genomic hybridisation
- congenital diaphragmatic hernia
- Fryns syndrome
- microdeletion
- submicroscopic chromosome deletion
Statistics from Altmetric.com
- array CGH, array comparative genomic hybridisation
- BAC, bacterial artificial chromosome
- CCD, charge-cooled device
- CDH, congenital diaphragmatic hernia
- CHR, Committee for Human Subjects Research
- CRL, crown-rump length
- FISH, fluorescence in situ hybridisation
- FS, Fryns syndrome
- PCR, polymerase chain reaction
- array comparative genomic hybridisation
- congenital diaphragmatic hernia
- Fryns syndrome
- microdeletion
- submicroscopic chromosome deletion
Fryns syndrome (FS; OMIM 229850) is the commonest autosomal recessive syndrome in which congenital diaphragmatic hernia (CDH) is a characteristic feature.1–3 FS comprises CDH and pulmonary hypoplasia, brachytelephalangy with nail hypoplasia, craniofacial dysmorphism, orofacial clefting, and organ malformations including cerebellar and neuronal heterotopias, ventricular septal defects, renal cysts, and bicornuate uteri.1–3 Diagnostic guidelines have been established but there are no formal diagnostic criteria for FS or currently known biochemical or molecular markers for FS, and the aetiology has not been determined.
Several different chromosome aberrations have been associated with a phenotype similar to FS.4,5 For example, Pallister-Killian syndrome or tetrasomy 12p has frequently been the cause of a presentation resembling FS with CDH, pulmonary hypoplasia, coarse facial features, aortic stenosis and cardiac septal defects, anal abnormalities, and hypoplasia of the external genitalia.6 Exclusion of this karyotype by skin biopsy has been recommended prior to making the diagnosis of FS. CDH is associated with an underlying cytogenetic abnormality in 10–33% of cases.4 Recurrent chromosome deletions have been shown to cause CDH and other phenotypic abnormalities.5 Deletions of chromosome 15q24 to 15qter have been associated with CDH from nine reports involving either “pure” monosomy for 15q24-15qter7–9 or monosomy for 15q24-15qter with additional chromosome imbalance.10–12 The critical region for CDH has been localised to chromosome 15q26.1 to 15q26.2 based on G-banded karyotyping.9 Clinical features in addition to CDH in 15q25-15qter monosomy have included congenital heart disease with hypoplastic left heart syndrome and coarctation of the aorta, pulmonary hypoplasia, reduced growth, mild facial dysmorphism, fifth finger clinodactyly and brachydactyly, talipes, and a single umbilical artery.7–9 Recurrent deletions of chromosome 8p23-8pter have also been associated with CDH, cardiac malformations, growth retardation, developmental delay, facial dysmorphism, and genitourinary anomalies.13–16 Heart defects have been found in at least 60% of cases with 8p23 monosomy and include atrio-ventricular canal defects, atrial septal defects, and ventricular septal defects.15 The diaphragmatic defects occur less frequently than the heart malformations and one review found that the 8p23 deletion phenotype included CDH in 4/18 cases.13 The critical chromosome band for CDH has not been molecularly defined, although one report established that chromosome band 8p23.1 was consistently deleted in probands with CDH.5
We are currently using array comparative genomic hybridisation (array CGH) to detect submicroscopic chromosome aberrations in patients with CDH and additional anomalies consistent with FS. We present three probands who were found to have submicroscopic chromosome deletions involving chromosome 15q26.2 and chromosome 8p23.1 that were not detected with G-banded karyotypes.
CASE REPORTS
The clinical features pertaining to the first two probands have been summarised in table 1. The first child was born to a 27 year old mother. Prenatal diagnosis revealed a left sided CDH, hypoplastic left heart syndrome, and talipes. Polyhydramnios was not detected. The baby was born at 39 weeks of gestation by C-section for fetal distress. She was apnoeic and cyanotic and the Apgar scores were two at 1 min and nine at 5 min. An echocardiogram revealed severe hypoplasia of the left heart and aortic arch and care was withdrawn shortly after birth because of the cardiac hypoplasia. External examination showed a female infant with weight 1800 g (<10th centile), crown-to-heel length of 41.5 cm (<10th centile), and a head circumference (OFC) of 30 cm (<10th centile). The head was normocephalic and no dysmorphism was reported (fig 1). A cleft palate was identified. The external examination was otherwise normal apart from mild talipes equinovarus. The umbilical cord had two vessels. Internal examination showed a large, left sided posterior diaphragmatic hernia with herniation of the liver and bowel into the thoracic cavity and displacement of the heart to the right side of the chest. There was marked hypoplasia of the left ventricle with stenosis of the mitral and aortic valves. The aortic arch was hypoplastic with aortic coarctation, but the right ventricle and pulmonary and tricuspid valves were normal. The left lung weighed 1 g and had two hypoplastic lobes and the right lung weighed 7 g and had three lobes (measurements consistent with severe pulmonary hypoplasia). The morphology of the other internal organs was unremarkable. Chromosome analysis from a sample of skeletal muscle showed an apparently normal female karyotype (46, XX) at 400 band resolution. A diagnosis of FS was suspected during the pregnancy, but no syndrome diagnosis was made after birth.
Previously reported cases with “pure” monosomy for 15q24-15qter and the first two probands

Facial photograph of the first proband.
The second child was the first baby born to a 33 year old mother. A prenatal ultrasound scan showed multiple abnormalities including a diaphragmatic hernia and cardiac anomalies. Polyhydramnios was not detected. A prenatal diagnosis of trisomy 13 was suspected, but the parents declined genetic amniocentesis. Preterm labour started at 32 weeks of gestation and a female infant was delivered with Apgar scores of five at 1 min and one at 5 min. Birth weight was 2153 g (>90th centile), crown-rump length (CRL) was 31 cm (<10th centile), and OFC was 31 cm (50–75th centile). The infant died shortly after delivery. External examination (fig 2) showed aplasia cutis with a 1.5 cm scalp defect near the vertex, and widened fontanelles. Hypotelorism was described but the only measurement available was the outer canthal distance at 6.6 cm (25th centile). The nasal bridge was broad and flat, and the ears were low set. There was a cleft palate and micrognathia. The extremities showed ulnar deviation with a flexion contracture of the right hand and narrow nails. There was webbing of the neck and hirsutism was present on the back. The umbilical cord had two vessels.

Photograph of the second proband showing craniofacial dysmorphism.
Internal examination showed a large, left sided CDH with herniation of the spleen and bowel into the left pleural space. There was pulmonary hypoplasia and the right lung weighed 1.8 g and the left lung 0.8 g (expected total combined weight: 34 g (SD 11 g)). The heart showed transposition of the great vessels with marked enlargement of the pulmonary artery and a small and rudimentary aorta. The aortic valve was stenotic with a third, misshapen semilunar valve. There was a large atrial septal defect, a single ventricular cavity with an absent ventricular septum, and a patent ductus arteriosus. There was bilateral hydroureter and distal atresia of the ureters prevented drainage of urine into the bladder (fig 3). The kidneys had dilatation of the calyces with thinning of the cortices and there was a double uterus and a double septate vagina with normal fallopian tubes and ovaries. Microscopic examination showed hepatosplenomegaly with erythroid hyperplasia in the liver and spleen consistent with reduced oxygen supply to the fetus. Chromosome testing on cord blood showed a normal female karyotype at a 500–600 band resolution (46, XX). A diagnosis of FS was made after birth.

Photograph from the second patient at autopsy showing ureteral atresia and double vagina and uterus.
The third proband was the third child born to a 33 year old mother. A sonogram was performed for a positive triple screen result (Down syndrome risk of 1 in 160) and showed left sided CDH, an atrial septal defect, ascites and pleural effusions, and polyhydramnios. Labour was induced at 22 weeks of gestation and a stillborn male (fig 4) was delivered with a weight of 586 g, OFC of 22 cm, and CRL of 18.5 cm (all measurements appropriate for gestational age). An autopsy showed complete absence of the left hemidiaphragm with severe pulmonary hypoplasia, incomplete lobation of the right lung, dysmorphism with a broad nasal bridge and micrognathia, a large atrial septal defect and a significant ventriculoseptal defect, malrotation of intestines, and a micropenis with cryptorchidism (normal for gestation). There was hypoplasia of the fingernails and toenails. Chromosome analysis on amniocytes showed a normal male karyotype (46, XY) and fluorescence in situ hybridisation (FISH) studies for chromosome 22q syndrome were negative. A diagnosis of FS was made at autopsy.
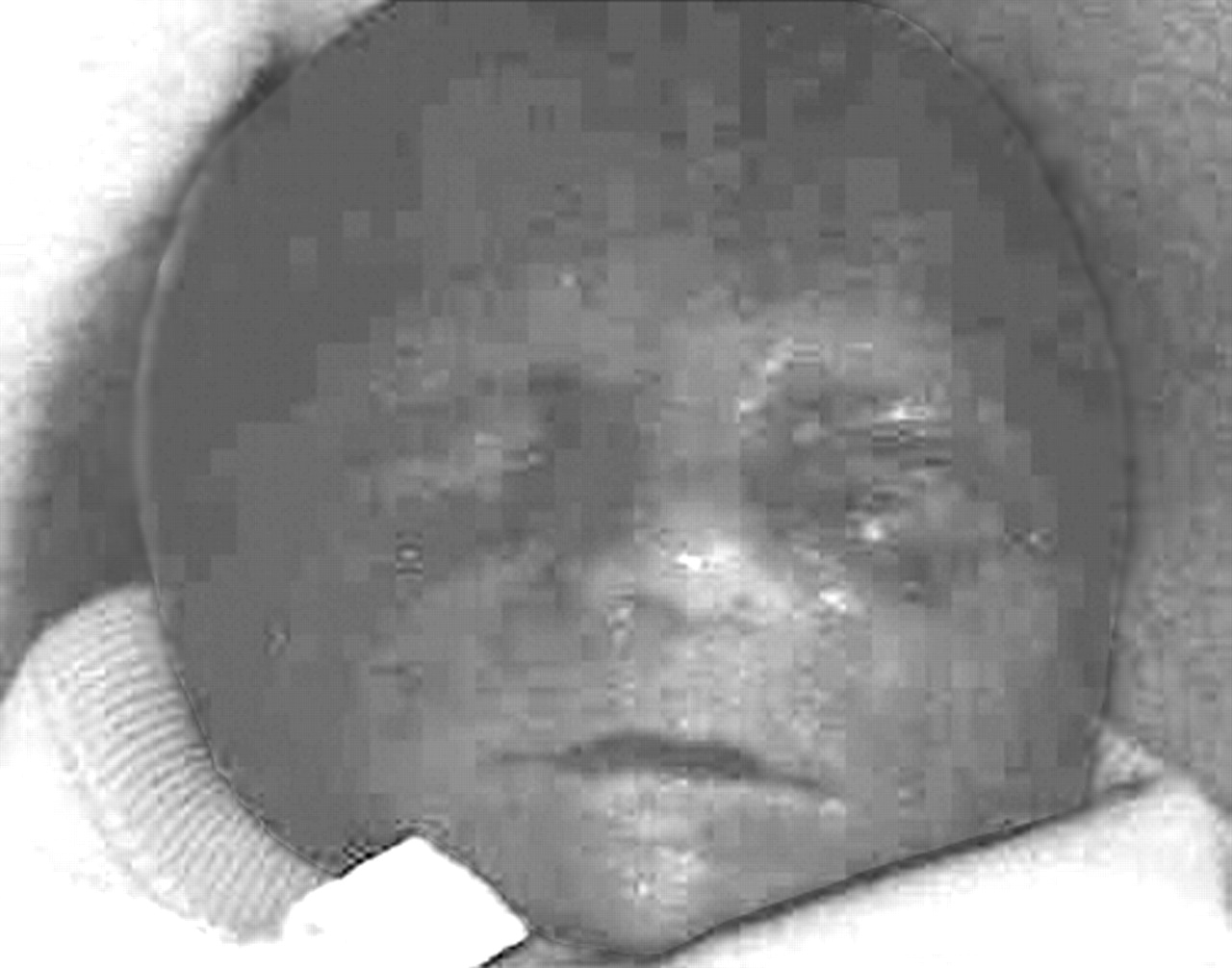
Facial photograph of the third proband, showing a broad nasal bridge and micrognathia.
METHODS
DNA samples were obtained from probands and parents under a protocol approved by the Committee for Human Subjects Research (CHR) at the University of California, San Francisco (UCSF; CHR number H41842-22157-02A). DNA was extracted from peripheral blood lymphocytes from the parents or from paraffin sections from the probands by digestion with proteinase K and salting out according to standard protocols. Array CGH was performed on DNA extracted from paraffin sections from the three probands to identify copy number differences between the probands and normal controls. The HumArray 2.0 bacterial artificial chromosome (BAC) array comprising 2464 BAC, PAC, and P1 clones was used17 and was obtained from the UCSF Comprehensive Cancer Center Microarray Shared Resource (http://cc.ucsf.edu/microarray). The majority of the clones (2442/2464; 99%) on the array are single copy with an average resolution of 1.4 Mb.17 The array CGH methodology has previously been described.17,18 Briefly, patient DNA and reference DNA were labelled by random priming with fluor-conjugated nucleotides and unincorporated nucleotides were removed using spin columns. The labelled DNA was mixed with an excess of Cot-1 DNA, precipitated, and redissolved in hybridisation buffer containing 50% formamide, 10% dextran sulphate, 2×SSC, and 2% SDS. The DNA was denatured at 72°C for 10 min and preannealed at 37°C for 1 h. After hybridisation at 37°C for 16–72 h, slides were washed with 50% formamide, 2×SSC, and 0.1% SDS at 45°C followed by 0.1 M sodium phosphate buffer with 0.1% Nonidet P-40. Images of the DAPI, Cy3, and Cy5 fluorescence intensities were collected using a charge-cooled device (CCD) camera system19 and analysed by software programs including SPOT and SPROC.20 The ideal log2 ratios for heterozygous genomic deletions encompassing a whole BAC are −1 for a single copy deletion and +0.58 (or less) for single copy duplications.17
Microsatellite marker analysis was used to verify the array CGH data for the probands, as cell lines were unavailable for FISH studies. The initial marker screens were performed with markers spaced at 5 cM intervals from a high density marker set (HD5; ABI Prism linkage mapping set v2.5). Polymerase chain reaction (PCR) was performed in 96 well plates and the PCR products were mixed with formamide, loading dye, and size standards. The products were denatured and separated using an ABI Prism 3700 DNA analyser (Applied Biosystems, Foster City, CA) and the markers were multiplexed according to marker size and fluorescent dyes. Analysis of the electropherograms was performed using GeneMapper 3.1 software (Applied Biosystems). Allele sizes were assigned using two reference individuals from CEPH families with known genotypes. Following the initial screen, additional 15q26 markers were selected (table 2).
Microsatellite markers at chromosome15q26.1 to 15qter in the first two probands
RESULTS
In the first patient, array CGH showed reduced copy number for two BAC clones at 15q26.2 consistent with monosomy for this chromosome region (data not shown). The two hybridisation spots with reduced log2 ratios values less than −1 were BAC clones RP11-185D5 (ratio −1.167) and RP11-9B2 (ratio −1.059) at chromosome 15q26.2. We considered that the array result together with previous reports of CDH and monosomy for 15q24-15qter and the phenotypic similarity of the proband to previously reported patients (table 1) were sufficient to justify further study of this chromosome region in this patient. Microsatellite marker analysis showed that the proband has not inherited a paternal allele for markers D15S207, D15S1014, and D15S212 mapped to 15q26.2 (table 2; fig 5A). Markers D15S130 and marker D15S966 were uninformative, and thus deletion size was estimated to be between 7 and 14 Mb in size.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Microsatellite marker D15S1014 in the proband’s mother (top line), father (second line), and proband (third line). The proband has inherited a single allele from his mother but no allele is present from the father. (B) Microsatellite marker D15S533 in the proband’s mother (top line), father (second line), and proband (third line). The proband has inherited a single allele from his father but no allele is present from the mother. (C) Microsatellite marker D8S550 in the proband’s mother (top line), father (second line), and proband (third line). The proband has inherited one allele from his father, but no allele is present from the mother.
In the second patient, array CGH showed reduced copy number for BAC clones RP11-185D5 (ratio −0.751), RP11-9B21 (ratio −0.87), and GS25P4 (ratio −0.848) at 15q26 to 15qter, consistent with a deletion for this chromosome region (data not shown). In view of the findings in the previous proband, we performed microsatellite marker analysis that showed that the proband had not inherited a maternal allele for marker D15S533, mapped to 15q26.2 (table 2; fig 5B). Flanking marker D15S212 was uninformative and markers D15S1014 and D15S120 were inherited in a Mendelian pattern, consistent with an interstitial deletion of 1–2 Mb estimated size.
In the third proband, array CGH showed reduced copy number for five BAC clones at 8p22-23.1, implying possible haploinsufficiency for this chromosome region (data not shown). The five hybridisation spots with reduced log2 ratios with values less than −0.85 were BAC clones RP11-112G9 (ratio −0.958), RP11-241I4 (ratio −0.893), RP11-23505 (ratio −0.967), RP11-235I5 (ratio −0.867), and RP11-262B7 (ratio −0.829) at chromosome 8p23.1 (data not shown). We considered these results suggestive of a deletion at 8p22-8p23, and microsatellite marker analysis showed that the proband had not inherited a maternal allele for markers D8S503, D8S550, and D8S520 mapped to chromosome 8p23.1 (fig 5C). The estimated size of this deletion ranged from 2 to 6 Mb (table 3).
Microsatellite markers at chromosome 8q23.1 for the third proband
In the first and third probands, the presence of two alleles for each parent with markers that are deleted in their child is evidence that the deletions are de novo. In the family of the second child, maternal haploinsufficiency for marker D15S533 as found in the proband has not definitely been excluded, although if present, this has not been associated with any of the phenotypic effects noted in the proband. These results also show that array CGH and microsatellite markers can successfully be used to identify submicroscopic chromosome deletions in probands who are deceased when only paraffin sections are available.
DISCUSSION
We have identified three infants who had CDH and physical anomalies suggestive of FS with submicroscopic chromosome deletions using array CGH after initial G-banded karyotyping had yielded normal results. In the first proband, FS was considered as a diagnosis before birth and in the second and third probands, FS was diagnosed following delivery. These results show that phenotypes similar to FS can be caused by submicroscopic chromosome deletions in cases in which conventional chromosome analysis has reportedly been normal. We would therefore recommend high resolution karyotyping as a mandatory investigation in patients suspected of having FS, with consideration of further studies with array CGH if possible to exclude submicroscopic cytogenetic abnormalities. The finding of two different microdeletions suggests genetic heterogeneity for phenotypes similar to FS, although the aetiology of FS remains unknown. FS has been considered to have autosomal recessive inheritance because of parental consanguinity21–24 and it seems premature to conclude that karyotypic abnormalities will be the sole cause of this phenotype. However, chromosome aberrations may constitute a greater fraction of the aetiology of cases with CDH and additional malformations than previously suspected.4
In the first two female probands, submicroscopic deletions of chromosome 15q26 were demonstrated using microsatellite marker analysis and in the third proband, a deletion of chromosome 8p22-8p23 was identified. The phenotypic findings in the first two infants were consistent with previously reported cases of “pure” 15q26-15qter monosomy and both had CDH, pulmonary hypoplasia, heart defects, a cleft palate, and a single umbilical artery (table 1). In particular, the CDH and cardiac defects in these two probands were similar to previous cases, with one reported infant having hypoplastic left heart syndrome8 and another reported infant having abnormalities of the aortic valve.9 However, the second proband in this report also had additional physical anomalies including a double uterus and vagina and ureteral atresia, despite a smaller chromosome deletion than the first proband. The explanation for these further physical findings is unclear and the array CGH did not definitively allow the exclusion of other chromosome aberrations, although the finding of an interstitial 15q26.2 deletion in combination with the patient’s phenotype suggests that the CDH and other physical findings are likely to have been caused by the deletion. The two microdeletions in these patients overlap but were not identical and were inherited on different parental alleles, making it unlikely that imprinting contributed to the phenotype. We propose that deletions of 15q26.2 constitute a new and recognisable gene deletion syndrome that includes the findings of CDH, pulmonary hypoplasia, cardiac malformations with hypoplastic left heart syndrome and aortic valve abnormalities, craniofacial dysmorphism, cleft palate, genitourinary anomalies, talipes, and a single umbilical artery. The frequency of chromosome deletions at 15q26 in patients with CDH and congenital heart disease remains to be determined. The CDH and congenital heart disease are likely to be caused by haploinsufficiency for a gene or genes in this chromosome region, although it is plausible that the deletions could alter the expression of a closely positioned gene. Candidate genes at 15q26 include MEF2A, a member of the myocyte specific enhancer factor-2 (MEF2) family that enhances the differentiation of mesodermal precursor cells to myoblasts and is involved in cardiac myocyte development.25 There are three characterised genes in the 15q26.2 chromosome region, MCTP2, N2RF2 (OMIM 107773), and MGC44294. Of these, NR2F2 (nuclear receptor sub family2, group F member 2; also known as COUP-TFII) has been the best studied and is known to be involved in angiogenesis and cardiac development.26 Targeted deletion of this gene in mice has resulted in embryonic lethality and the mutant mice are unable to model primitive capillary plexus to microcapillaries; in addition, the atria and sinus venosus do not develop past the primitive heart tube stage.26 However, none of these genes has so far been shown to be associated with a Fryns syndrome-like phenotype.
In the third proband, a microdeletion of chromosome 8p22-8p23 was demonstrated by microsatellite analysis and the proband had pulmonary hypoplasia and cardiac defects with a large atrial septal defect and a ventricular septal defect. These findings are consistent with previous cases with monosomy for chromosome band 8p23.1 that have had CDH, pulmonary hypoplasia, complex congenital heart defects and atrioventricular canal defects, facial dysmorphism, and varying degrees of mental retardation.13 The gata4 gene is involved in cardiac morphogenesis in the mouse and the human homologue maps to 8p23.1-8pter.16GATA4 haploinsufficiency has been demonstrated in patients with monosomy 8p and congenital heart disease, but not all patients with heart defects have had deletions including this gene.27 In this patient, the GATA4 gene is not definitely deleted as the gene is positioned between markers D8S550 (deleted in patient 3; table 3) and D8S552 (not deleted in patient 3; table 3). No critical interval for the gene(s) causing the CDH associated with 8p23 monosomy has been molecularly defined.
CONCLUSION
We have demonstrated submicroscopic chromosome deletions at chromosome 15q26.2 to 15qter in two females and a submicroscopic chromosome deletion involving 8p23.1 in a male. All three had CDH and additional anomalies consistent with FS. We conclude that CDH and a presentation similar to FS can be caused by a new, clinically identifiable chromosome deletion syndrome at 15q26.2 comprising CDH, pulmonary hypoplasia, congenital heart disease, craniofacial dysmorphism including cleft palate, genitourinary malformations, talipes, and a single umbilical artery. We also conclude that submicroscopic chromosome deletions should be excluded whenever possible in patients with CDH and a phenotype similar to FS.
ACKNOWLEDGEMENTS
We are grateful to Ms Melanie dela Cruz and Mr Jack Malek for technical assistance.
REFERENCES
Footnotes
-
This work was supported by REAC grant number 36248-524269, University of California, San Francisco. We also acknowledge the Sandler Family Supporting Foundation for a Sandler Program in Basic Science/New Technology Resources Award to the Genomics Core Facility.
-
Competing interests: none declared
-
Ethics approval: The protocol used in this study was approved by the Committee for Human Subjects Research (CHR) at the University of California, San Francisco (UCSF; CHR number H41842-22157-02A).
Statement regarding photographic reproduction: Signed permission for reproduction of photographs was obtained from a parent for each of the probands under protocol CHR number H41842-22157-02A at UCSF.
Since the submission of this paper, additional case reports of diaphragmatic hernia associated with deletions at chromosome 15q26 have been published (
;). A 5 Mb “critical interved” has been delineated for the diaphragmatic hernia causing gene by Klaasens et al between BACs RP11-152L20 and RP11-753A21 (2005).