Article Text

Abstract
Background: BRCA1 associated RING domain protein (BARD1) was originally identified due to its interaction with the RING domain of BRCA1. BARD1 is required for S phase progression, contact inhibition and normal nuclear division, as well as for BRCA1 independent, p53 dependent apoptosis.
Methods: To investigate whether alterations in BARD1 are involved in human breast and ovarian cancer, we used single strand conformation polymorphism analysis and sequencing on 35 breast tumours and cancer cell lines and on 21 ovarian tumours.
Results: Along with the G2355C (S761N) missense mutation previously identified in a uterine cancer, we found two other variants in breast cancers, T2006C (C645R) and A2286G (I738V). The T2006C (C645R) mutation was also found in one ovarian tumour. A variant of uncertain consequence, G1743C (C557S), was found to be homozygous or hemizygous in an ovarian tumour. Eleven variants of BARD1 were characterised with respect to known functions of BARD1. None of the variants appears to affect localisation or interaction with BRCA1; however, putative disease associated alleles appear to affect the stability of p53. These same mutations also appear to abrogate the growth suppressive and apoptotic activities of BARD1.
Conclusions: These activities allowed us to identify one of the rare variants (A2286G; I738V) as a neutral polymorphism rather than a detrimental mutation, and suggested that G1743C (C557S) is not a polymorphism but may contribute to the cancer phenotype.
- ANN, axillary node negative
- BARD, BRCA1 associated RING domain
- LOH, loss of heterozygosity
- SSCP, single strand conformation polymorphism
- BARD1
- BRCA1
- mutations
- apoptosis
- tumour suppressor
Statistics from Altmetric.com
- ANN, axillary node negative
- BARD, BRCA1 associated RING domain
- LOH, loss of heterozygosity
- SSCP, single strand conformation polymorphism
Breast cancer will affect one woman in eight at some point in her lifetime, of whom <10% have inherited a predisposition to breast cancer, and of those, less than half have a mutation in the BRCA1 or BRCA2 gene (see references in Sauer1). The others are probably due to mutations in other moderate to high penetrance genes. Indeed, several mutations have previously been identified in the BRCA1 associated RING domain gene, BARD1,2 in non-BRCA1/2 hereditary site-specific breast and breast/ovarian cancer cases.3–5 The vast majority of breast cancers, however, are sporadic in nature. BRCA1 and BRCA2 are rarely found mutated in sporadic breast cancers. In contrast, both germline and somatic BARD1 mutations have been reported in sporadic breast, ovarian, and uterine cancers.6 Some of these BARD1 variants are of unclear clinical relevance; several result in amino acid substitutions that would be expected to have a major effect, yet they have been denoted as polymorphisms; others are predicted to be tolerated alterations based on evolutionary conservation, but are disease associated; and there is disagreement over whether one (C557S) is truly disease associated or a polymorphism.3,5,6
BARD1 encodes a 777 residue protein with an aminoterminal RING domain (residues 46–90), three ankyrin repeats (residues 427–525) and two carboxyterminal BRCT domains (residues 616–653 and 743–777); it also has a nuclear export signal (residues 102–120)7 and a nuclear localisation signal (after residue 177, potentially residues 204–209) (supplemental fig 1, available from the JMG website at http://www.jmedgenet.com/supplemental; reviewed in Irminger-Finger & Leung8). It can form stable heterodimers with BRCA1,9,10 facilitated by residues adjacent to the RING domain (residues 26–119).11–13BRCA1 tumour associated mutations in the RING domain appear to disrupt the BARD1 interaction and stabilisation in vivo, but there are no known BARD1 RING domain mutations.11,14

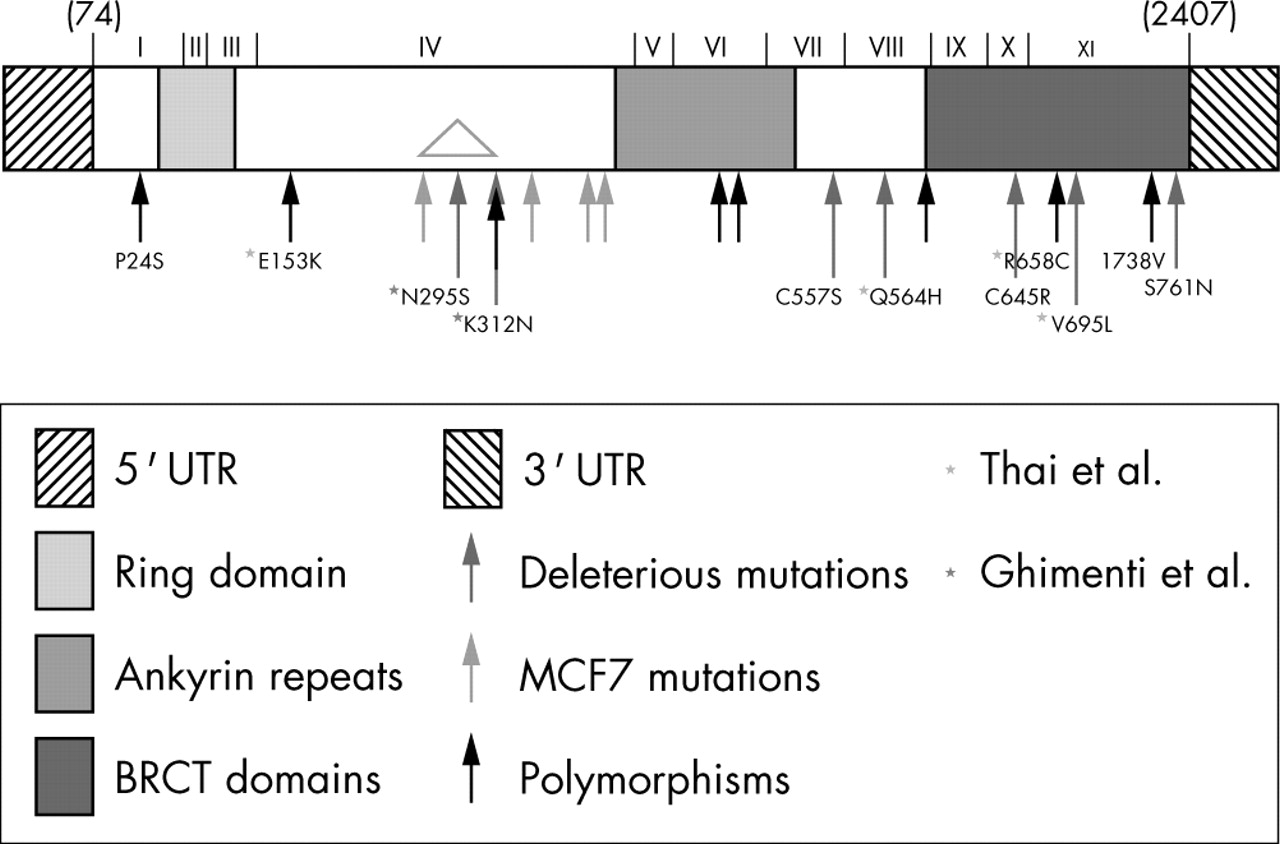
Schematic showing location of missense alterations in BARD1. Both protein domains and cDNA exons are indicated (adapted from Thai et al. Hum Mol Gen: 1998; 7: 195–202). Black arrows designate polymorphisms, blue arrows designate mutations found in MCF7 and orange arrows designate deleterious mutations. Purple asterisks indicate mutations identified only by Thai et al, while green asterisks indicate mutations identified only by Ghimenti et al. Variants used for characterisation are named.
BARD1 is required for S phase progression, contact inhibition and normal nuclear division, alterations consistent with up regulation of cell cycle inhibitors.15,16 It is also required for apoptotic response to genotoxic stress, mediating apoptosis in a BRCA1 independent, p53 dependent manner.17,18 BARD1 colocalises with BRCA1 and RAD51 in S phase nuclear dots. Its RING domain is an E3 ubiquitin ligase that is more active when present as a heterodimer with the BRCA1 RING domain;19–26 tumour derived RING domain mutations in BRCA1 disrupt this activity. Although autoubiquitination occurs in vitro, it does not appear to result in the degradation of BRCA1 or BARD1. The carboxyterminal region of BARD1 (including the final two ankyrin repeats and the BRCTs) associates with CstF50 and inhibits polyadenylation.27,28 The BARD1 germline mutation resulting in Q564H reduces binding of BARD1 to CstF and abrogates the inhibition of polyadenylation.28 This region is also involved in binding to the Ewing’s sarcoma protein, EWS, modulating its transrepression and transactivation activities,29 and binds to the Bcl3 ankyrin repeats, forming complexes on NFkB binding sites to modulate transcription.30
We present here the results of genetic analysis of BARD1 in a set of 31 breast tumours, 21 ovarian tumours, and 6 cell lines. We have identified 10 missense alterations, 4 silent changes and 1 deletion/insertion; 9 of these have not been previously reported. In order to determine the effect, if any, of the missense alterations on the function of BARD1, we examined 11 variants in transient culture assays to measure their effect on growth inhibition, apoptosis and associations between BARD1, BRCA1, and p53.
MATERIALS AND METHODS
Specimens
Unselected breast tumours were obtained from our large prospectively accrued (Toronto, Ontario, Canada, 1987–1993) cohort of axillary node negative (ANN) cases.31 Tumour tissue was snap frozen upon harvesting and stored in liquid nitrogen until processed. cDNAs made from mRNA isolated from 31 quick frozen, sporadic, node negative tumours and from four cell lines (MCF7, MDA 231, MDA 468, and T47D) were analysed. For the ovarian cancer samples, DNA was extracted from paraffin embedded ovarian tumour sections as described.32 Genomic DNAs from 21 ovarian tumours and from four cell lines (MCF7, MDA468, and SKBR3 breast cancer lines; SW480 colon cancer) were analysed. As controls, genomic DNAs isolated from the blood of 45 healthy individuals were analysed. Tumour samples were obtained with patient consent and were encoded to preserve anonymity.
PCR-SSCP analysis
PCR was performed in 20 ul or 25 ul volumes containing 1 ul cDNA (from 25 ng mRNA) or 100 ng genomic DNA, respectively, using the primer pairs and conditions given in table 1 (see table 1 in supplemental data, available from the JMG website at http://www.jmedgenet.com/supplemental) and 1 μCi α33P-dATP and 0.2 μl AmpliTaq DNA polymerase (Roche). Cycling conditions were: denaturation at 94 °C for 3 minutes; 35 cycles of 94 °C for 15 seconds, annealing for 15 seconds (see table 1 in supplemental data), and 72 °C for 30 seconds; and a final extension at 72 °C for 10 minutes. Samples were diluted 1:3 in formamide buffer, denatured at 95 °C for 3 minutes, then rapidly cooled on ice for 3 minutes, following which 2.5 ul of each sample was loaded onto a 5% single strand conformational polymorphism (SSCP) gel in 0.5× TBE buffer, and electrophoresed at 9 W (8–10 W) overnight in a 4 °C cold room. Gels were dried and then exposed to film for 24–48 hours.
BARD1 variants
Sequencing
For all samples exhibiting shifts, the PCR was repeated without radioisotope, and the gel products were purified and subjected to manual cycle sequencing using α33P-dATP thermosequenase (Invitrogen) and one of the external primers. Cycling conditions were: 40 cycles of denaturation at 94 °C for 10 seconds, annealing (at appropriate temperature) for 10 seconds and extension at 72 °C for 15 seconds. Loss of heterozygosity (LOH) was determined from sequencing gels; the presence of the wild type sequence along with a variant sequence suggested no LOH.
Construction of BARD1 expression vectors
The BARD1 gene was subcloned into pFLAG-CMV6. This vector can be used in transient expression assays and encodes FLAG-epitope fusion proteins so that the exogenous proteins can be differentiated from endogenous BARD1. The variant alleles were created by the two step PCR mutagenesis method.33 All mutagenised regions were sequenced to ensure that no other mutations had been inadvertently introduced by Taq polymerase. In total, 11 variant alleles were constructed in this manner: 7 putative disease associated alleles (A957G (N295S), A1009T (K312N), G1743C (C557S), G1756C (Q564H), T2006C (C645R), G2156C (V695L), and G2354A (S761N)), and 4 putative benign polymorphisms for comparison (C143T (P24S), G530A ((E153K), C2045T (R658C), and A2285G (I738V)).
Proliferation assay
Assays were performed using the Roche cell proliferation kit (MTT) protocol. Cells were seeded at approximately 103cells/well in 100 ul of culture media with or without 10 umol/ml muristerone in flat-bottomed 96 well dishes. pIND (Invitrogen) constructs were transfected using Fugene-6 into a SKBR3 derived cell line previously selected in Zeocin for inducibility with muristerone (due to expression of the receptor plasmid, VgRxR). Stable cell lines inducibly expressing the pIND constructs were doubly selected in neomycin and Zeocin. All time points were performed in triplicate and averaged.
Colony assays
NIH3T3, HEP293T or SKBR3 cells were cotransfected with 1 ug of each pCMV-Flag-BARD1 allele along with 1 ug of a puromycin resistance vector. The pCMV-Flag vector was used as a negative control. Transfections were performed using Fugene-6 (Roche). Cells were trypsinised after 1 day and an aliquot grown in Puromycin for approximately 2 weeks before scoring.
Apoptosis assays
NIH3T3 cells were transiently transfected with 1 ug of each of the Flag-BARD1 alleles under the control of the CMV promoter. The vector alone was used as a negative control. Transfections were performed using Fugene-6 (Roche), with efficiencies of 10% to 25% (as determined by transfections with RFP-BARD1 or GFP, data not shown). Cleaved PARP (Asp214) antibody fluorescein conjugate (Cell Signaling) and annexin-V-FLUOS (Roche) assays were performed as described by the manufacturers.
p53 immunoprecipitation and Western blotting
HEP293T cells were transiently cotransfected with 3 ug of each pCMV-Flag-BARD1 allele and 1 ug of pcDNA3.1-p53 using Fugene-6 (Roche). Cell lysates were analysed by immunoprecipitation with monoclonal anti-FLAG M2 affinity gel (Sigma) or polyclonal anti-p53 agarose gel (FL-393; Santa Cruz) and Western blot analysis with monoclonal anti-FLAG antiserum (M5; Kodak) or monoclonal anti-p53 antiserum (1C12; Cell Signaling). The antibodies were used at 1 ug/ml. HRP conjugated rabbit anti-mouse IgG was used at 1/5000 for Western blot analysis.
RESULTS
Mutation analysis of BARD1
We analysed cDNAs from 35 breast tumours and breast cancer cell lines, and genomic DNA from 21 ovarian tumours and 4 cancer cell lines by SSCP analysis of the entire coding region. Sequencing of the SSCP variants identified seven as missense alterations that each occurred a single time, one (T2006C (C645R)) that occurred in a breast and an ovarian tumour, and one (G1592A (V507M)) that was present in four samples (table 1, fig 1). We also identified four silent changes, which occurred in between one and five samples each.
Two missense (C143T (P24S) and G1592A (V507M)) and two silent (C1126G (T351) and C1591T (H506)) alterations have previously been classified as polymorphisms4,6 (although the G1592A (V507M) variant was associated with an increased risk of breast cancer in postmenopausal women4). Another missense alteration (G2355A (S761N)) had previously been identified as a somatic mutation with associated LOH in a uterine cancer.6 The missense alteration (G1743C (C557S)) was previously labelled as a polymorphism by one group6 but as a disease associated germline mutation by other groups.3,5 Unlike the published reports, our sample showed evidence of LOH—that is, it appeared to be hemizygous. The two remaining novel missense alterations (A2285G (I738V) and T2006C (C645R)) needed further analysis to determine whether they were detrimental alleles, although neither showed evidence of LOH.
In an analysis of 45 normal blood samples, no shifts were observed using the primers for the T2006C (C645R) or the A2285G (I738V) variant, nor were these variants found in the remainder of the cancer samples. The other variants were not tested for, as they had previously been identified and classified as polymorphisms or disease associated alterations in other publications.4,6
Characterisation of missense alterations in BARD1
To characterise these missense alterations epidemiologically would require a large number of samples and matched controls; a better approach would be to combine available genetic data with functional studies. We therefore characterised a set of 11 variant alleles (see fig 1; from our work and published reports) in functional assays to determine if the mutations were neutral or deleterious.
BARD1 is a tumour suppressor gene, and thus is expected to cause a decrease in growth rate if ectopically expressed. Indeed, inducible expression of wild type BARD1 in a stable SKBR3 derived (breast cancer) cell line resulted in cessation of cell growth (fig 2). As a means of rapid analysis of growth suppression, the 11 variant alleles were used in transient colony assays in SKBR3, 293T, and NIH3T3 cell lines. They were cotransfected with a puromycin resistance plasmid, and the relative number of drug resistant colonies compared with empty vector cotransfections was determined. Equal amounts of plasmid DNAs were used and, as seen in fig 3, expression levels of the different variants were approximately the same. Transfection with the wild type allele, or with the putative neutral polymorphisms (encoding P24S, E153K, R658C, and I738V), resulted in a reduced number of puromycin resistant colonies; assays performed using the putative disease associated alleles (encoding N295R, K312S, C557S, Q564H, C645R, V695L, and S761N) resulted in numbers of drug resistant colonies more similar to that seen with the vector control than wild type (table 2).
Puromycin-resistance colony assay

Expression of BARD1 results in inhibition of cell proliferation. Proliferation assays comparing inducible expression of wild type BARD1 and empty vector are shown. MTT absorbance units are proportional to number of live cells present at that time point. Each datapoint was performed in triplicate and averaged. Navy diamonds indicate no inducer was added to the cells while pink squares indicate cells induced with muristerone. Top: SKBR3-4:pIND5, a stable SKBR3-derived cell line expressing the inducible vector pIND alone; bottom: SKBR3-4:BARDmp, a stable SKBR3 derived mixed population cell line expressing the inducible BARD1wt allele. Representative proliferation assays are shown.

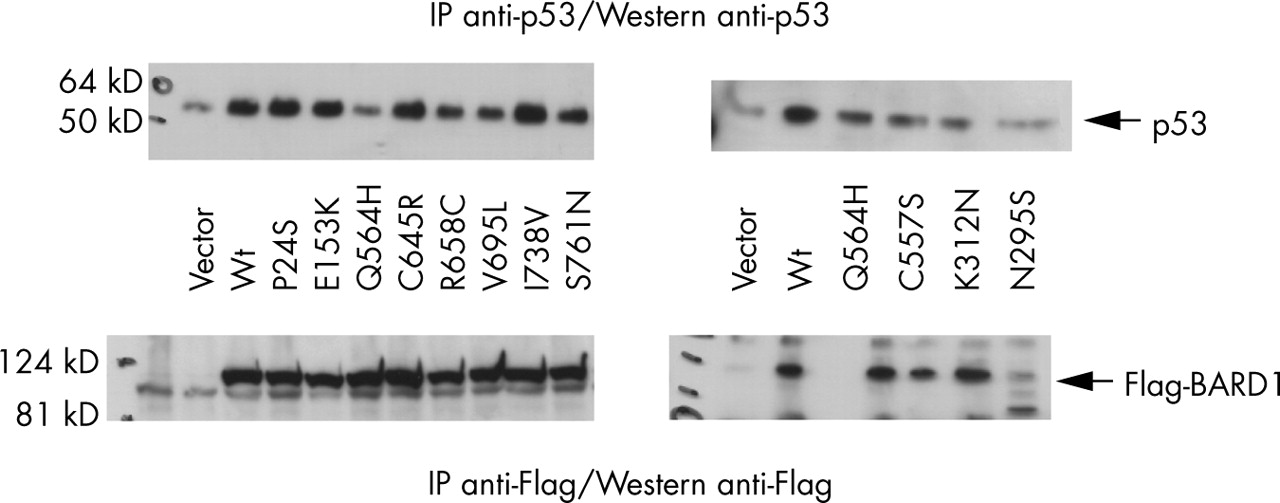
BARD1 variants differentially affect p53 stability. Transiently transfected HEP293T cell lysates were split in half; one aliquot was analysed by immunoprecipitation and Western blot analysis using anti-p53 antisera (upper panels), the other using anti-Flag antisera (lower panels). The two right hand panels are from a single experiment; the two left hand panels are from a different experiment. Both were repeated. Arrows indicate the p53 bands (upper panels) or Flag-BARD1 bands (lower panels). Lower levels of p53 protein are seen in lysates from cells transfected with the empty vector or with the deleterious mutants encoding Q564H, R658C, V695L, S761N, C557S, K312N, and N295S, yet BARD1 protein levels are the same in all samples. However, note the cleavage product for the BARD1 N295S variant. Wt, wild type allele.
These data were supported by results from apoptosis assays (table 3). Transfection with the wild type allele or the neutral polymorphisms resulted in approximately 25% PARP positive cells (equivalent to the transfection efficiency using a GFP expression plasmid; data not shown) and strong annexin V positivity. In contrast, transfection with the vector or the putative disease associated variants resulted in little or no evidence of apoptosis. A defect in induction of apoptosis was seen previously for the Q564H variant.17
Apoptosis assays
The roles of BARD1 in growth suppression and apoptosis are probably linked to its association with the tumour suppressor p53. We found evidence that wild type BARD1, along with the neutral polymorphic variants, stabilised p53 (fig 3); p53 levels were higher in lysates with exogenous expression of the wild type and polymorphic alleles than in lysates with exogenous expression of the deleterious alleles. The levels of the exogenous BARD1 variant proteins were approximately equivalent in all cell lysates, with the exception of the N295S variant; this protein appears to be less stable, with a major cleavage product at approximately 80 kD (compared with 110 kD for the wild type).
In contrast, co-immunoprecipitation assays with BRCA1 and BARD1 showed no differences between the neutral polymorphisms and the putative disease associated alterations (data not shown). Similarly, none of the missense alterations characterised here had any effect on the cellular localisation of the encoded proteins—that is, they all demonstrated a punctate nuclear staining similar to that seen with the wild type allele34 (supplemental fig 2 available from the JMG website at http://www.jmedgenet.com/supplemental).
DISCUSSION
BARD1 appears to be a high penetrance but low abundance cancer predisposition gene. Unlike BRCA1, it is found mutated in a small percentage of both sporadic (our data and those of Thai et al6) and non-BRCA1/2 familial breast or breast/ovarian cancers.3,4 We, and others, have identified seven missense alterations in breast, ovarian, and uterine cancers, which encode deleterious variants: N293K, K312N, C557S, Q564H, C645R, V695L, and S761N. Other missense variations, and presumably the silent changes, are likely to be neutral polymorphisms.
Alterations occur throughout the BARD1 gene, with no obvious hot spots. Several disease associated variants (C645R, V695L, and S761N) result in alterations within the BRCT domains; two occur between them and the ankyrin repeats (C557S and Q564H), while others (N295S and K312N) result in alterations outside of known structural or functional domains. Several tumour derived mutations in BRCA1 also occur within the BRCT domains, suggesting that these perform some essential functions in both of these proteins. No mutations have been found to date in the RING domain, unlike for BRCA1, suggesting that BARD1 may have a role(s) in tumour progression independent of its interaction with BRCA1.
If effects of the various missense alterations on the functions of the BARD1 protein were to be predicted a priori, based only on the amino acid substitutions, many would probably be misclassified. Even computer prediction programs such as PolyPhen (Polymorphism Phenotyping; http://www.bork.embl-heidelberg.de/PolyPhen) and SIFT (Sorting Intolerant from Tolerant; http://blocks.fhcrc.org/sift/SIFT.html) were correct for only 3/7 and 2/7, respectively, of the disease associated missense alterations, and 4/5 and 1/5, respectively, of the neutral polymorphisms. Both programs only correctly identified I738V as a benign polymorphism, yet of two similar substitutions, with equivocal computer results, one (V507M) appears to be a polymorphism while the other (V695L) appears to be cancer associated. Thus, classification of variant alleles as polymorphic versus deleterious is not straightforward, particularly for those identified in non-hereditary cases.
Instead, we used functional assays to classify a set of missense alterations as benign polymorphisms or deleterious variants. As a tumour suppressor, ectopic expression of BARD1 is expected to cause a decrease in cell growth; this was indeed seen for induction of the wild type allele. In the transient colony assays, there is a correlation of loss of growth suppression with the putative disease associated/deleterious variants encoding N293K, K312N, C557S, Q564H, C645R, V695L, and S761N, and no loss of function in the putative benign polymorphisms encoding P24S, E153K, R658C, and I738V. These results were seen even more clearly in the transient apoptosis assays. It should be noted that C557S and Q564H map within the minimal region required for BARD1 apoptosis.18
Our assay results are consistent with reports classifying G1743C (C557S) as a deleterious variant3,5 rather than a polymorphism. Although no definitive explanation can be put forth here for the findings of Thai et al6 that this variant occurred twice in their samples from a white control population, it is clear from the protein assays that this is a non-functional allele; the allele may not be fully penetrant, or the “healthy” individuals may not yet have presented with cancer.
BARD1 probably acts via p53; there is a correlation between the growth suppression and apoptotic capabilities of the BARD1 variants and the levels of p53 protein in cell lysates. Functional BARD1 appears to stabilise p53, which in turn promotes apoptosis as well as cell cycle inhibition. The exact mechanism for this is not addressed here, although others have reported a direct interaction between BARD1 and p5317, and a requirement of the BRCA1/BARD1 complex as a scaffold for the ATM/ATR dependent phosphorylation of p53.10 Although the RING domain of BARD1 (and BRCA1) is an E3 dependent ubiquitin ligase,9,19–21,35 it does not appear to cause the degradation of p53, as seen by the increased levels of p53 in cells transfected with wild type and polymorphic alleles of BARD1. Along with BRCA1, it may function, instead, to activate or potentiate p53 activity in response to DNA damage.
The absence of mutations within the RING domain/BRCA1 association region is the likely reason for the lack of correlation between BRCA1 binding and stability and BARD1 mutational status. None of the missense alterations appears to have an effect on the cellular localisation of the encoded proteins, and as they also have no observable effect on the interaction of BARD1 and BRCA1, it is expected that BRCA1 protein will be correctly localised to the nucleus in S phase dots. This, in keeping with the report that BARD1 has a BRCA1 independent, p53 dependent role in apoptosis,8,17 suggests that BARD1 has a role in tumorigenesis separate from its association with BRCA1, and that the missense alterations found in the deleterious alleles affect those BARD1 specific function(s).
Acknowledgments
We would like to thank M Soares for her work on the BARD1 mutation analysis in breast tumours and J Kwan for her work on the BARD1 mutations analysis in ovarian tumours.
REFERENCES
Supplementary materials
The table is available as a downloadable PDF (printer friendly file).
If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
- [view PDF] - Table: PCR primers and conditions for amplification of BARD1from cDNA or genomic DNA templates


{kind=link}
{kind=link}
{kind=link}
Footnotes
-
This study was supported by a Terry Fox Program Project Grant, a grant from the NCI to the Ontario site of the Cooperative Family Registry for Breast Cancer Studies (U01 CA69467) and by Cancer Care Ontario.
-
Competing interests: none declared
-
All necessary ethics committee approvals were secured for this study.