Article Text

Abstract
Patients with hereditary haemorrhagic telangiectasia (HHT, or Osler-Weber-Rendu syndrome) have variable presentation patterns and a high risk of preventable complications. Diagnostic tests for mutations in endoglin (HHT type 1) and ALK-1 (HHT type 2) are available. Some HHT patients are now known to have HHT-juvenile polyposis overlap syndrome due to Smad4 mutations. Families were ascertained following the presentation of probands for embolization of pulmonary arteriovenous malformations. Genome-wide linkage studies using over 700 polymorphic markers, and sequencing of candidate genes, were performed. In a previously described HHT family unlinked to endoglin or ALK-1, linkage to Smad4 was excluded, and no mutations were identified in the endoglin, ALK-1, or Smad4 genes. Two point LOD scores and recombination mapping identified a 5.4 cM HHT3 disease gene interval on chromosome 5 in which a single haplotype was inherited by all affected members of the pedigree. The remainder of the genome was excluded to a 2–5 cM resolution. We are currently studying a further family potentially linked to HHT3. We conclude that classical HHT with pulmonary involvement can result from mutations in an unidentified gene on chromosome 5. Identification of HHT3 should further illuminate HHT pathogenic mechanisms in which aberrant transforming growth factor (TGF)-β signalling is implicated.
- AVMs, arteriovenous malformations
- CM-AVM, capillary malformation-arteriovenous malformation
- EBV, Ebstein-Barr virus
- EC, endothelial cells
- HBT, hereditary benign telangiectasia
- HHT, hereditary haemorrhagic telangiectasia
- HHT1, HHT type 1
- HHT2, HHT type 2
- JP, juvenile polyposis
- JPHT, juvenile polyposis/HHT overlap syndrome
- R-Smads, receptor-associated Smad proteins
- TGF-β, transforming growth factor-β
- ALK-1
- arteriovenous malformations
- endoglin
- juvenile polyposis
- TGF-β
Statistics from Altmetric.com
- AVMs, arteriovenous malformations
- CM-AVM, capillary malformation-arteriovenous malformation
- EBV, Ebstein-Barr virus
- EC, endothelial cells
- HBT, hereditary benign telangiectasia
- HHT, hereditary haemorrhagic telangiectasia
- HHT1, HHT type 1
- HHT2, HHT type 2
- JP, juvenile polyposis
- JPHT, juvenile polyposis/HHT overlap syndrome
- R-Smads, receptor-associated Smad proteins
- TGF-β, transforming growth factor-β
Hereditary haemorrhagic telangiectasia (HHT, also known as Osler-Weber-Rendu syndrome) is one of the most common autosomal dominant disorders, affecting between 1 in 5000 and 1 in 8000 people in Europe and Japan.1,2 HHT is a genetically heterogeneous group of disorders that lead to common vascular phenotypes. HHT types 1 and 2 have been recognised for more than a decade. HHT1 (OMIM 187300) results from mutations in endoglin3 on chromosome 9, whereas the disease gene for HHT2 (OMIM 600376) is ALK-1 on chromosome 12.4 In addition, mutations in Smad4/MADH4 causing a juvenile polyposis/HHT overlap syndrome (JPHT; OMIM 175050) have been described recently.5 Although the existence of a third “pure” HHT locus has been suggested twice,6,7 the first family were subsequently demonstrated to have an ALK-1 mutation,8 and further data on the family described by Wallace and Shovlin7 have not been presented.
All forms of HHT result in the development of abnormal blood vessels including telangiectasia of the oral mucous membranes, nose, and gastrointestinal tract, and visceral arteriovenous malformations (AVMs). Nosebleeds and chronic gastrointestinal bleeding leading to iron deficiency anaemia and transfusion dependence are the features of HHT most appreciated by clinicians. Visceral AVMs are usually silent, but screening programmes indicate that pulmonary AVMs occur in 30–50% of HHT patients,9,10 cerebral AVMs in 10%,9 and hepatic AVMs in 20–30%.6,11 Pulmonary AVM-induced embolic strokes and brain abscesses, and cerebral AVM-induced haemorrhagic strokes make HHT a common cause of inherited stroke in young adults,12 and complications from other visceral involvement, including hepatic failure, also occur. There are subtle differences in the phenotype between HHT1 and HHT2, with HHT2 patients exhibiting fewer pulmonary AVMs and a milder HHT phenotype,13 but carrying a higher risk of development of HHT related pulmonary arterial hypertension14 (table 1).
HHT genes and pattern of HHT
Importantly, many of the complications of HHT can be prevented or limited by clinical screening programmes. Since HHT is a disease with late onset penetrance (≈90% by 40 years; 97% by 60 years15), genetic screening programmes have been introduced.16 Patients without detectable mutations in endoglin or ALK-1 are recognised by the HHT genetic centres. It would be predicted that some of these, particularly from smaller families will have Smad4 mutations since routine colonoscopies that would exclude juvenile polyposis are not a feature of HHT management. In this group, there would be additional clinical screening implications, since for at risk members of juvenile polyposis (JP) families, the British Society of Gastroenterology recommends surveillance colonoscopies and upper gastrointestinal endoscopies, with therapeutic interventions to reduce later risks of colon cancer.17
The pathogenic mechanisms involved in the development of the HHT vessels are of interest to scientists and clinicians alike. Endoglin and ALK-1 encode proteins expressed predominantly on vascular endothelial cells. Endoglin, ALK-1, and the ubiquitously expressed Smad4 are involved in signalling by the transforming growth factor (TGF)-β superfamily that regulates a diverse series of fundamental pathways in development and pathophysiology. A simplified model of Smad dependent signalling by this superfamily is presented in fig 1, indicating the interactions and functions of the HHT gene products. Ligands signal through heteromeric complexes comprised of type I and type II cell surface receptor serine-threonine kinases.18 Activated type I receptors phosphorylate cytoplasmic receptor associated Smad proteins (R-Smads). These oligomerise with a co-Smad molecule, Smad4, and translocate to the nucleus to act as transcription factors and alter gene expression.

TGF-β superfamily signalling in EC indicating roles of the known HHT gene products. Endoglin associates with all three groups of transmembrane signalling receptors27 and modulates TGF-β1 induced cellular responses.28 Expression of ALK-1 on EC allows TGF-β1/TβRII to transduce signals through Smad1 and Smad5, whereas in most cell types, TGF-β/TβRII initiate Smad2 and Smad3 signalling following activation of ALK-5.29 BMPs, bone morphogenetic proteins; GDF, growth/differentiation factors. Boxes: HHT gene products. *: gene excluded by linkage analyses.
In view of the clinical implications of the new molecular association of HHT with juvenile polyposis, we consider it important to report the linkage analysis in the classical HHT pedigree described by Wallace and Shovlin.7 This identifies a new HHT gene locus (HHT3) on chromosome 5, resulting in four known types of HHT (table 1).
METHODS
Pedigrees
The proband (fig 2, III.3) was referred to the Hammersmith Hospital for embolization of pulmonary arteriovenous malformations (AVMs). Extended pedigree analysis was performed with informed consent and Multicentre and Local Research Ethics Committee approval (MREC/98/0/42; LREC 99/5637M). The diagnosis of HHT was assigned by the presence of three international consensus diagnostic criteria,19 that is: affected first degree relative; recurrent, spontaneous nosebleeds; mucocutaneous telangiectasia; and in the case of III.3 and III.4, documented visceral manifestations (pulmonary AVMs). Importantly, telangiectasia were considered diagnostic only if in the correct distribution for HHT (that is, nose, lips, tongue, oral mucosa, finger tips, or ears) and persistent, having developed from late childhood or during adult life. In view of nosebleeds affecting 8–10% of children, with nocturnal nosebleeds a common feature,20 occasional nosebleeds occurring purely in childhood were not considered a diagnostic criterion.

Family pedigree and haplotype analysis. Pedigree: Individuals III.3 and III.4 have pulmonary AVMs. DNA from II.3, II.4, III.3, and III.4 was sequenced for candidate gene analyses. Haplotype analysis: Inheritance of 10 haplotypes in the pedigree indicates segregation of disease haplotype (outlined) in all affected individuals over four generations (II–V). Data on clinically unaffected individuals are not included for ethical reasons, and data on uninformative markers have been excluded for clarity. Key: [or]: recombination events; ( ): data deduced from offspring; -: data not available.
Genotyping and molecular analyses
Genomic DNA was extracted from peripheral venous blood or Isocode mouth swabs (Schleicher and Schuell, Dassel, Germany) using standard procedures. cDNA was derived from Ebstein-Barr virus (EBV) immortalised lymphocyte cell lines which were established on four family members as previously.21
A total of 400 fluorescently labelled primer pairs from the ABI Prism Linkage MD-10 Mapping Set (Applied Biosystems, Foster City, CA) were used according to the manufacturer’s instructions for a first genome-wide linkage screen. An additional 312 fluorescently labelled polymorphic markers pre-labelled from Applied Biosystems and Research Genetics (Huntsville, AL), or custom synthesised by Sigma-Genosys (Cambridge, UK), were used to fine-map the identified interval, and exclusion map the remainder of the genome. PCR products were size separated on a ABI 7700 capillary sequencer, and analysed using GeneScan software (Applied Biosystems). Candidate genes were analysed by PCR amplification of all exons, exon-intron boundaries, and 40–50 bp of flanking intronic sequence (endoglin, ALK-1, Smad4, Smad5), or by sequencing the entire cDNA from EBV immortalised lymphocyte cell lines (SPARC). Primer details are available on request.
Two point LOD scores between a putative disease locus and each marker were calculated assuming autosomal dominant inheritance, a disease gene frequency of 0.0001, and equal recombination rates in both sexes. LOD scores were calculated initially with equal allele frequencies. Based on previously published estimates,15,22 for unaffected individuals, penetrance was set at p = 0.8 between ages 12 and 35 and p = 0.95 aged over 35 giving two liability classes. Apparently unaffected children under the age of 12 years were excluded as penetrance is less than 80% at this age.22 SLINK and MLINK analyses23 were performed using Human Genome Mapping Project (HGMP) computational resources. The order and distances between the markers were derived from Ensembl (http://www.ensembl.org/) and GenBank (http://www.ncbi.nlm.nih.gov/) databases.
RESULTS
Exclusion of known HHT disease genes
Linkage analyses were performed to confirm the exclusion of endoglin and ALK-1 in the extended pedigree, and to exclude the new JP-HHT gene, Smad4 (table 2).
Exclusion of known HHT genes
Endoglin had been sequenced in full in affected members of the pedigree.21 The other two HHT genes were sequenced in four affected family members (III.3, II.3, II.4, III.3, and III.4). No mutations were found in any of the ALK-1 coding regions or intron-exon boundaries. Although the JP-HHT mutations predominantly occur in the 3′ exons of Smad4,5 all 11 exons and intron-exon boundaries were sequenced. No mutations were found.
Exclusion of core components of the TGF-β signalling pathways
Recognising that the data from the other HHT genes strongly suggested that the disease gene in this pedigree would encode a protein affecting TGF-β signalling, other core components of TGF-β signalling pathways were excluded by linkage analyses (see fig 1). Literature and database searches revealed that in addition to the proteins illustrated in fig 1, over 100 further proteins are known to interact with the TGF-β superfamily signalling pathways, precluding an exhaustive candidate gene approach. A genome-wide screen was therefore undertaken.
Linkage analyses define the HHT3 locus on chromosome 5
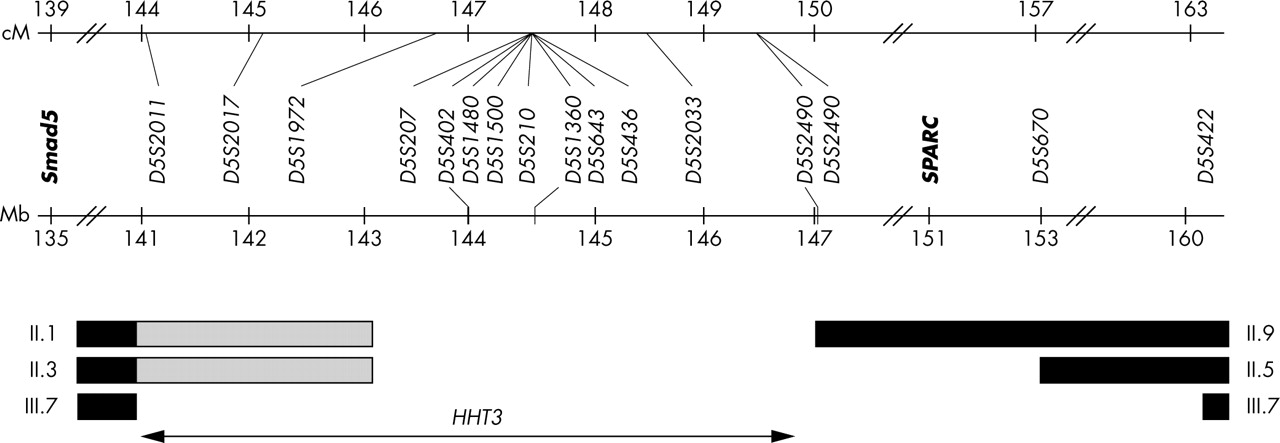
An initial genome-wide scan excluded 70% of the genome, and identified a 12 cM interval where LOD scores exceeded +2. Information from initial markers was limited due to non-informative meioses. Supplementary adjacent markers were fully informative, generating a two point Zmax of 3.45 at a recombination fraction (θ) of 0.00, and refining the interval. LOD scores were robust to changes in allele frequency (data not shown). The SLINK theoretical Zmax of 4.84 at θ = 0.00 was not achieved due to recombination events in clinically unaffected individuals. The series of two point LOD scores (table 3) and recombination mapping using affected individuals (fig 3) defined the 5.4 cM/6 Mb HHT3 locus. In this region, all affected family members had inherited a conserved disease associated haplotype (fig 2).
LOD scores (Z) spanning the HHT3 interval
{kind=link}
{kind=link}
{kind=link}
Recombination mapping of interval using affected family members. Pedigree numbers as in fig 2. Black bars indicate definite recombination events; shaded bars indicate uninformative markers. The locations of the candidate genes Smad5 and SPARC (but not RASA1 at 86.6 Mb) are illustrated.
In order to assess the likely frequency of HHT3, we studied three families (including two previously unreported) with theoretical Zmax>1.6 in which assignment to endoglin or ALK-1 could not be made. In two, linkage to HHT3 was excluded. In a third family with a theoretical Zmax of 1.87, a maximum two point LOD score of 1.17 at θ = 0.00 was obtained with D5S436 on a different disease-segregating haplotype to that illustrated in fig 2. The reduction from the theoretical Zmax was due to a single young unaffected recombinant.
Exclusion mapping
To exclude the possibility that an alternative locus had been missed, the remainder of the genome was formally examined. A further 290 polymorphic markers were selected and analysed to ensure that at least two double recombination events would have had to occur in a 2–5 cM interval for a putative locus to have been missed. Highly conservative estimates (excluding genetic interference) based on 500 intervals indicated that the probability of this occurring was between 3.1×10−3 and 8×10−5 (that is, p⩽0.0031).
Candidate gene analysis
Ensembl identifies 28 genes within the 5.4 cM HHT3 interval, including 10 of unknown function. Furthermore, the gene for Smad5, a strong candidate based on its role in ALK-1 signalling pathways (fig 1), is assigned on current mapping to within 5 Mb of the HHT3 interval (fig 3). The Smad5 gene had been excluded by linkage analyses using markers either side of the published gene locus. However, in view of its strong candidate status due to functional considerations, and the possibility that the precise database positional assignment of Smad5 was erroneous, all coding exons and flanking intronic sequence were sequenced in affected members of both families. No pathogenic mutations were identified. In addition, the initial 12 cM mapping interval contained SPARC, a further attractive candidate gene due to endothelial cell expression and roles in TGF-β1 mediated proliferative responses.24 SPARC cDNA was amplified from EBV immortalised lymphocyte cell lines. The complete transcript was sequenced and no mutations identified.
DISCUSSION
We have identified a novel locus for the autosomal dominant disorder hereditary haemorrhagic telangiectasia (HHT). In the presented family, the disease affects both sexes equally and is indistinguishable from that in other families with HHT. The pulmonary AVM frequency (13%) was not as high as in HHT type I families with endoglin mutations, but numbers are too small to suggest that HHT3 resembles HHT2 more than HHT1. Importantly, no family members have experienced cancer of any form, and none are known to have developed pulmonary hypertension.
Our data do not allow us to address the proportion of HHT families which are due to HHT3, as in our four “unassigned” large families, only one categorically maps to chromosome 5. Most HHT families will have mutations in endoglin or ALK-1, and mutational screening programmes should detect the majority of these. Data from labs employing sensitive quantitative genomic exon PCR screening methods have not detected mutations in as many as 10–15% of classical HHT families (Dr Michelle Letarte, personal communication). In these families, linkage analyses with the chromosome 5 markers should begin to address the likely frequency of HHT3.
The HHT3 locus (5q31.3–5q32) is not the same as that recently identified for hereditary benign telangiectasia (HBT; OMIM 187260) on chromosome 5 (5q14),25 for which the causative gene, RASA1, encoding Ras GTPase activating protein 1, has been identified.26 HBT is therefore part of the capillary malformation-arteriovenous malformation (CM-AVM) syndrome,26 and should not be considered a benign allelic variant of HHT as proposed.25 The importance of making this clear distinction is that HHT patients are at significant risk of pulmonary and cerebral AVMs. The diseases can be distinguished clinically by the distinctive skin lesions. In HBT, randomly distributed cutaneous vascular malformations over the head, trunk, and limbs are often congenital (40%) or develop from early childhood. In contrast, HHT telangiectasia have a highly restricted distribution on the mucosa of the nose, lips, oral cavity, conjunctiva, finger tips, ears, and face,19 develop from teenage years, and become more numerous with age.
As all three identified HHT genes encode proteins involved in TGF-β superfamily signalling, we anticipate that the disease gene responsible for HHT3 will also encode a protein involved in Smad dependent TGF-β signalling. In keeping with the expression patterns of endoglin and ALK-1 which are transmembrane proteins predominantly expressed on vascular endothelial cells (EC), we predict that the disease gene for this “pure” form of HHT will also display EC restricted expression. Identification of this gene will be important not only for clinical diagnostic services but also to elucidate the mechanisms of TGF-β superfamily signalling in vascular endothelium.
Acknowledgments
We are grateful to the families for their participation in these studies, the MRC-CSC Core Sequencing Facility at Hammersmith, Dr Carol Shoulders for provision of approximately 100 polymorphic markers, Dr Anna Marrone for assistance in the establishment of the EBV immortalised cell lines and Dr Julian Walters for discussions on juvenile polyposis phenotype and screening. We also thank Dr Carol Shoulders, Professor Anne Soutar, Dr Bernard Morley, and Dr Michael Jones for helpful discussions and draft manuscript reviews.
REFERENCES
Footnotes
-
This project was primarily funded by the British Heart Foundation, with additional support from the Margaret Hayton HHT Memorial Fund.
-
Competing interests: none declared