Article Text

Abstract
Background: Inactivating mutations of the gene RS1 lead to X-linked retinoschisis, a progressive retinal dystrophy characterised by schisis within the inner layers of the neuroretina. The mutation spectrum is large and the phenotype variable.
Aim: To determine whether there is a correlation between mutation type and disease severity.
Methods: We identified the causative mutation in 86 affected patients and examined each of these patients in detail. Different categories of mutation were compared for each phenotypic characteristic.
Results: We found a reduction in visual acuity with increasing age and worsening macular pathology in patients over 30 years old (p⩽0.001), but there was no correlation between mutation type and severity of disease. Furthermore, we found a wide variation in phenotype even within families.
Conclusions: Identifying the causative mutation in patients with X-linked retinoschisis is helpful in confirming diagnosis and in counselling of family members but cannot be used to predict prognosis for an individual patient.
- DVA, distant visual acuity
- FERG, full field electroretinogram
- NVA, near visual acuity
- XLRS, X-linked juvenile retinoschisis
- genotype
- phenotype
- retinoschisin
- retinoschisis
Statistics from Altmetric.com
- DVA, distant visual acuity
- FERG, full field electroretinogram
- NVA, near visual acuity
- XLRS, X-linked juvenile retinoschisis
X-linked juvenile retinoschisis or congenital retinoschisis (XLRS) is the leading cause of juvenile macular degeneration in males1 and is characterised by schisis (splitting) of the inner layers of the neurosensory retina resulting in characteristic foveal schisis.2–4
XLRS is fully penetrant1 but with considerable phenotypic variation.1,4–6 Peripheral retinal abnormalities are seen in 50–70% of patients7 and complications include vitreous haemorrhage, retinal detachment, and neovascular glaucoma.2,6 The full field electroretinogram (FERG) characteristically shows a normal a-wave with a reduced amplitude b-wave,8 although a-wave changes have been described in some patients.9
Heterozygous female carriers of RS1 mutations do not generally show clinical or ERG abnormalities,3 although one girl has been reported with foveal schisis and a reduced b-wave on FERG.10 Females who are homozygous for an RS1 mutation exhibit similar symptoms and signs to affected males.11,12
The causative gene RS1, in Xp22.1, encodes a 224 amino acid protein retinoschisin.13 Retinoschisin function is unknown, but its discoidin domain has high homology to proteins implicated in cell adhesion. Retinoschisin is found only in the retina,13 is secreted, and functions as an oligomer.14–16 The RS1 gene is expressed late in retinal development17 by neuronal cells within the retina and retinoschisin is present in both the photoreceptor and inner nuclear layers.15,18 Over 100 different mutations have been identified and are inactivating (X-linked Retinoschisis sequence variation database at http://www.dmd.nl/rs/ and Human Gene Mutation Database at http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html). The majority of these are missense mutations in exons 4–6 but also include nonsense and splice site mutations, deletions, and insertions.13,19–23 Many missense mutations have been shown to prevent secretion of the mutant protein leading instead to intracellular degradation.14
There have been reports comparing genotype and phenotype.24–29 No correlation has been found between mutation type and disease severity, but these studies have been limited by small patient numbers. We report here the results of our study of 86 patients with XLRS in whom the causative RS1 mutation has been identified.
METHODS
Patients
A total of 86 UK patients were included in the study. Each had previously been diagnosed with XLRS and all were re-examined by one of three ophthalmologists (DP, NDLG, ATM). Five had no known family history of XLRS, while the others came from 29 families with a history of XLRS. The causative mutation for each patient had either already been identified or was identified as part of the study. All patients gave consent to participate in the study.
Ophthalmological examination
The refractive error in each eye and best corrected distant and near visual acuities were recorded. Ocular motility and pupil reactions were assessed, anterior segment examination performed with a slit lamp, intraocular pressures measured, and posterior segment examination performed with indirect ophthalmoscopy and slit lamp biomicroscopy, following pupil dilation. FERG recordings were obtained in 43 of the 86 patients and were consistent with a diagnosis of XLRS.
Genetic studies
In a proportion of the patients studied the mutation had already been identified as part of a previous study.19 For the other patients the causative mutation was identified as previously described.19
Statistical methods
The age of the patients was recorded at the time of clinical examination. Five characteristics were chosen to describe the phenotype of each patient: (i) best corrected visual acuity, (ii) best corrected near vision, (iii) presence of foveal schisis or other foveal pathology in one or both eyes, (iv) peripheral schisis in one or both eyes, and (v) complications (vitreous haemorrhage or retinal detachment).
RS1 mutations were categorised depending on (i) whether they affected a conserved amino acid residue, (ii) whether they affected a residue within the discoidin domain, secretory leader peptide, or the remaining part of the gene, (iii) whether they affected a cysteine residue, (iv) the exon in which they occurred, (v) the mutation type: protein truncating or missense, (vi) whether the mutations interfered with protein secretion or allowed secretion into the extracellular space, and (vii) if successfully secreted, whether the mutations interfered with oligomerisation.
Each mutation category was compared to each phenotypic characteristic, using the χ2 test of independence. Phenotypes of patients from the same family and phenotypes of unrelated patients with the same mutation and of similar age, were compared.
For comparison of age and visual acuity, data were compared with those of a previous study.2 We used linear regression using the log transformed visual acuity and compared these data sets with an analysis of covariance, where we allowed for age as a linear covariate and tested for a difference in slopes by testing the interaction between age and study.
RESULTS
A total of 86 patients fulfilled the inclusion criteria and the diagnosis was supported by the FERG findings for 43 of these. Genotype details are summarised in table 1.
Genotype details of patients
Phenotype
The age range was 2–69 years with a distribution skewed towards a younger age (mean age 23.8 years, median 34.5 years). A total of 25 (29%) of our patients were younger than 10 years, 23 (27%) were between 11 and 20, 11 (13%) were between 21 and 30, and only 27 were older than 30 years (31%) (fig 1A).

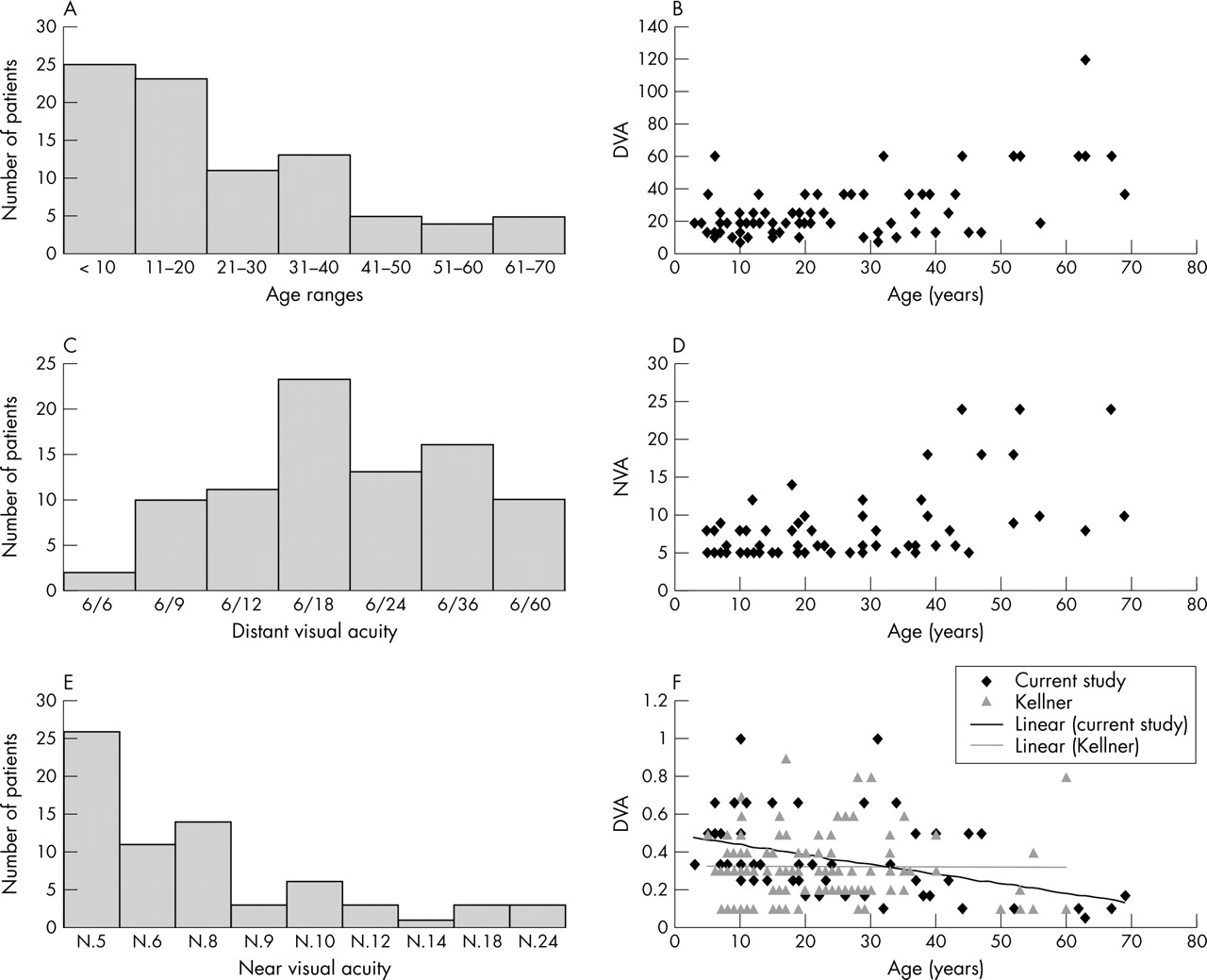{kind=link}
Age ranges and visual acuity for patients with X-linked retinoschisis. (A) Age range of 86 patients in the study. (B) Distant visual acuity (DVA) for each patient plotted as the denominator (6/X) against patient’s age. (C) The variation in DVA shown for the group of patients. (D) Near visual acuity (NVA) measured with vocational near vision test type for each patient plotted against age. (E) The variation in NVA shown for the group of patients. (F) A comparison of DVA for patients in this study compared to data from an earlier study.2 DVA is expressed as a decimal fraction, that is 6/60 = 0.1, to ease comparison with the earlier study.2 The key indicates the two datasets and regression lines are shown for each.
Distant (DVA) and near (NVA) visual acuity were measured for the patients (fig 1B–D). DVA varied from 6/6 to 6/60 and NVA from N.5 to N.24. Visual acuity appears to worsen with age over the whole group, but there is a large variation in severity with a patient only 5 years old with a DVA of 6/60 and another 47 years old with a DVA of 6/12. We found no correlation between visual acuity and complications or degree of peripheral schisis.
All patients had evidence of maculopathy. Forty eight patients (56%) had typical bilateral foveal schisis, 18 (21%) had foveal schisis in one eye and other maculopathy in the fellow eye, and 18 patients (21%) had other maculopathy in both eyes. Two patients had dense unilateral cataracts, not allowing fundal view of one eye, but had typical foveal schisis in the fellow eye. Patients over 30 years of age had more severe macular changes (p⩽0.001) which may explain the decrease in visual acuity with age.
Bilateral peripheral retinoschisis was detected in 42 patients (49%). Fifteen patients (17%) had peripheral schisis in one eye and either no schisis or retinal detachment in the fellow eye. No peripheral schisis was found in the eyes of 27 (31%) patients. There was no difference in the proportions of patients at a younger or older age with evidence of peripheral schisis. Retinal detachment or vitreous haemorrhage had occurred in 26 patients (30%) and affected both eyes of 11 patients and only one eye of 15. The proportions of younger and older patients who developed complications were similar.
A previous study of retinoschisis had reported no change in visual acuity with age in patients without complications.2 Sixty of our 86 patients had no complications and in this group we still found an association of reduced visual acuity with increasing age suggesting that visual acuity decreases with time (linear regression on log transformed data, p = 0.0013) perhaps due to a worsening of macular pathology.
Genotype
The causative mutation had been determined for each patient (table 1). Based on our previous molecular analysis of mutations in this protein,14,16 we were able to predict whether a particular missense mutation would lead to intracellular retention and degradation of retinoschisin or would allow secretion into the extracellular space (table 1). Only five mutations (seen in 18 patients) were predicted to lead to protein truncation.
We compared the clinical characteristics of groups of patients with similar mutations but found no evidence for a correlation between mutation type and disease severity even in patients of similar ages. We also compared visual acuity, foveal changes, peripheral schisis, and disease complications in patients with similar or different classes of mutations. We classified mutations into groups including protein truncating, missense, mutations in different exons, discoidin domain mutations and those affecting other residues, and mutations affecting cysteine residues and those affecting other residues. We found no significant correlation between mutation type and disease severity. We hypothesised that there may be a difference in disease characteristics between those patients predicted to have retinoschisin present within their retinal extracellular space (that is, mutant protein with a missense change but still allowing secretion) and those predicted to have no secreted protein (truncating mutations and missense changes shown to cause intracellular degradation of protein14,16). However, again, we found no significant difference in any of the clinical measures of disease severity.
Intrafamilial variation and phenotypic variation in patients of similar ages with the same mutation
We reviewed the phenotypic characteristics in families where we had access to large number of affected males. For two families we were able to examine six affected relatives and found the severity of disease was still very variable. We also compared young boys with the same mutation and again found a great deal of variation, for example, in boys with the R102W missense mutation originating from different families the visual acuity varied from 6/9 (11 year old) to 6/24 (10 and 14 year old).
DISCUSSION
The availability of genetic testing for families with X-linked retinoschisis has allowed accurate diagnosis and also improved genetic counselling for patients and relatives. Women at risk of being a carrier for this disorder can now be offered molecular genetic testing to determine whether they have an RS1 mutation. Sons of women who carry the mutation can be offered mutation testing to determine whether they will develop the disease.
Molecular genetic testing allows the specific mutation to be identified and could be used to predict long term prognosis if there is a relationship between disease phenotype and type of mutation. There have been several studies of the phenotypic variation in X-linked retinoschisis and comparisons made between patients with the same mutation24,26 and in groups of patients with different mutations,25 but as these studies are small limited conclusions can be drawn. We have studied a large group of patients with XLRS in whom the mutation has been identified and have related the mutations to our current knowledge about the molecular mechanisms thought to contribute to disease. We found a wide variation in clinical phenotype, both between and within families, which was not dependent on mutation type. Retinoschisin is a secreted protein thought to function within the extracellular retinal environment in cell adhesion.18 Many missense mutations lead to intracellular retention of the mutant protein leading to an absence of extracellular retinoschisin.14 However the severity of the disease in such cases is no different to those with mutations allowing secretion of retinoschisin. This, together with the variation seen within in families, suggests there are other factors influencing disease severity such as genetic modifiers (for example, genetic variation in interacting proteins) or environmental influences.
Molecular genetic testing in XLRS has facilitated diagnosis in atypical cases and has improved genetic counselling for families but unfortunately is of no value in predicting long term visual prognosis.
REFERENCES
Footnotes
-
We are indebted to Janice and John Pownall and Perkin-Elmer for charitable support. The study benefited from funding from the Medical Research Council, the Wellcome Trust, and the Guide Dogs for the Blind Association. DP was a Wellcome Trust Ophthalmology Training Fellow and NDLG was supported by the Guide Dogs for the Blind Association.
-
Competing interests: none declared