Article Text

Abstract
Background: Sotos syndrome (MIM 117550) is characterised by learning difficulties, overgrowth, and a typical facial appearance. Microdeletions at 5q35.3, encompassing NSD1, are responsible for ∼10% of non-Japanese cases of Sotos. In contrast, a recurrent ∼2 Mb microdeletion has been reported as responsible for ∼50% of Japanese cases of Sotos.
Methods: We screened 471 cases for NSD1 mutations and deletions and identified 23 with 5q35 microdeletions. We investigated the deletion size, parent of origin, and mechanism of generation in these and a further 10 cases identified from published reports. We used “in silico” analyses to investigate whether repetitive elements that could generate microdeletions flank NSD1.
Results: Three repetitive elements flanking NSD1, designated REPcen, REPmid, and REPtel, were identified. Up to 18 cases may have the same sized deletion, but at least eight unique deletion sizes were identified, ranging from 0.4 to 5 Mb. In most instances, the microdeletion arose through interchromosomal rearrangements of the paternally inherited chromosome.
Conclusions: Frequency, size, and mechanism of generation of 5q35 microdeletions differ between Japanese and non-Japanese cases of Sotos. Our microdeletions were identified from a large case series with a broad range of phenotypes, suggesting that sample selection variability is unlikely as a sole explanation for these differences and that variation in genomic architecture might be a contributory factor. Non-allelic homologous recombination between REPcen and REPtel may have generated up to 18 microdeletion cases in our series. However, at least 15 cannot be mediated by these repeats, including at least seven deletions of different sizes, implicating multiple mechanisms in the generation of 5q35 microdeletions.
- MLPA, multiplex ligation dependent probe amplification
- NAHR, non-allelic homologous recombination
- 5q35
- NSD1
- Sotos
- microdeletions
- overgrowth
Statistics from Altmetric.com
Sotos syndrome (OMIM 117550) is characterised by a typical facial appearance, learning difficulties, and overgrowth in childhood. Additional features include neonatal jaundice and hypotonia, cardiac and renal anomalies, scoliosis, seizures, and tumours.1–3 Sotos syndrome is caused by mutations and deletions of NSD1, a histone methyltransferase implicated in transcriptional regulation, which is located at chromosome 5q35.4–7
We previously reported that NSD1 intragenic mutations cause ∼76% of UK cases of Sotos syndrome, whereas microdeletions encompassing NSD1 cause ∼10% of cases.5 Similar results were reported for cases of Sotos from France, Germany, Italy, and the USA.6–9 However, intragenic mutations were reported in only 12% of Japanese cases of Sotos, whereas a recurrent ∼2 Mb microdeletion encompassing NSD1 was identified in ∼50%.10 It has been suggested that these contrasting results could be attributed to differences in sample selection, as the Japanese microdeletion cases did not all meet the stringent criteria for classic Sotos syndrome.
In this study we screened 471 cases with varying degrees of overgrowth and/or phenotypic overlap with Sotos syndrome for NSD1 mutations and deletions, to further evaluate the 5q35 microdeletion frequency in the UK and the possible reasons for differences between Japanese and non-Japanese populations. We performed microsatellite analyses in 33 cases with an NSD1 microdeletion to establish the size, parental origin, and mechanism of generation of the deletions, and undertook “in silico” analyses to identify sequence elements that may mediate the deletions.
SUBJECTS AND METHODS
Cases
The research was approved by the London Multicentre Research Ethics Committee, and consent was obtained from participating cases and/or parents. Through analyses of 471 cases ascertained through the Childhood Overgrowth Collaboration, 23 cases with microdeletions were identified. These 471 cases included ∼200 with a clinical diagnosis of Sotos syndrome or a Sotos-like syndrome. However, the majority of cases either had overgrowth and/or macrocephaly but did not have the facial gestalt of Sotos syndrome, or had facial features similar to Sotos syndrome, but no overgrowth. All 471 cases were screened for mutations and whole gene deletions of NSD1 as previously described.5 Fifteen microdeletion cases identified in these analyses were from the UK, and three (COG025, COG044, COG070) have been previously published.5 The remaining cases were from the Republic of Ireland, Australia, the USA, and France. Patients COG508a and COG508b were identical twins. NSD1 mutations were identified in 146 of the 471 cases.
We also ascertained microdeletion cases from published reports. Six cases were from France (COG342, COG343, COG344, COG345, COG346, COG3476) three from the USA (COG183, COG184, COG3998) and one from Italy (COG5489). DNA was obtained from both parents for 19 cases, from one parent for three cases, and was unobtainable for the remaining 11 cases. DNA from grandparents and/or siblings was obtained for 11 cases.
All cases with a microdeletion had been clinically diagnosed with Sotos syndrome prior to the molecular analyses. Clinical details were obtained in all cases except COG179, and photographs were available for 21 cases. These were independently assessed by five of us (TC, HEH, IKT, NR, and KT-B) and all were considered typical of Sotos syndrome.
Identification and delineation of 5q35 microdeletions
Microdeletions were identified and confirmed using at least two separate methods. Cases ascertained from the literature were identified by fluorescent in situ hybridisation using at least one intragenic NSD1 probe.6,8,9 The microdeletions were confirmed by multiplex ligation dependent probe amplification (MLPA) using the SALSA P026 NSD1 test kit and the methods described in Schouten et al.10 This kit contains probes for NSD1 exons 1, 2, 3, 5, 6, 11, 14, 17, and 23, FGFR4 exon 2, and 17 control probes. Microdeletion cases ascertained through the Childhood Overgrowth Collaboration were initially identified by analysis of the microsatellite marker, SOT3.5 This intragenic marker is highly polymorphic and within intron 2 of NSD1. Cases homozygous at SOT3 were further investigated with quantitative fluorescent PCR as previously described5 and identified microdeletions were confirmed using MLPA.
To determine the size of the microdeletions, polymorphic microsatellite markers within and surrounding NSD1 in cases and parental DNA were analysed. We had previously published four of these markers, SOT1, SOT12, SOT3, and SOT19,5 and we developed new markers using the UCSC Human Genome Project Working Draft sequence. The 2 Mb region encompassing NSD1 was searched for dinucleotide, trinucleotide, and tetranucleotide repeat elements and amplifying primers were designed using Primer 3 software. Twelve markers were informative and worked reliably in the analyses (Appendix 1). We also used three known microsatellite markers, D5S400, D5S2008, and D5S2073, and one FGFR probe from the SALSA P026 NSD1 kit. The position of the 20 markers relative to NSD1 is shown in figs 1A and 2. The forward primer for each marker was labelled with γ[32P]-ATP, amplified using PCR and the product was electrophoresed on a denaturing polyacrylamide gel. The gels were exposed to x ray film and the positions of the product bands were scored relative to each other.
To determine the parental origin and mechanism of generation of microdeletions we analysed 12 microsatellite markers from chromosome 5q35 in DNA from cases, parents, grandparents, and siblings. The order and distance between these markers was cen-D5S436-17 Mb-D5S422-6 Mb-D5S400-2.9 Mb-D5S429-3.7 Mb-SOT30-1 Mb-SOT27-0.3 Mb-SOT1-0.2 Mb-SOT3-1 Mb-D5S2008-19 kb-SOT23-1.6 Mb-D5S2073-1.2 Mb-D5S2006-tel. Haplotypes of marker alleles were determined for each family.
In silico analyses to identify and characterise repetitive elements flanking NSD1
Bioinformatic analyses were based on the May 2004 human genome sequence assembly, accessed at UCSC. To identify low copy repeat elements within the 3 Mb region surrounding NSD1, we divided the sequence into 2 kb fragments and compared them to one another using the BLAT program12 and a Perl script. Segment pairs with >90% sequence similarity over more than 200 bp were considered for further analysis, and plotted onto a 1500×1500 sequence similarity matrix, where duplicated regions clearly appeared as diagonals. To confirm that these duplicated regions were low copy repeats, the analysis was repeated with genomic sequence pre-masked for high copy repeats with RepeatMasker. The web based BLAT server at UCSC was used to validate these results and to obtain the percentage of sequence identity over the duplicated regions. The high copy repeat content of the NSD1 region was calculated with RepeatMasker using default settings.
RESULTS
NSD1 is flanked by three homologous low copy repeat elements
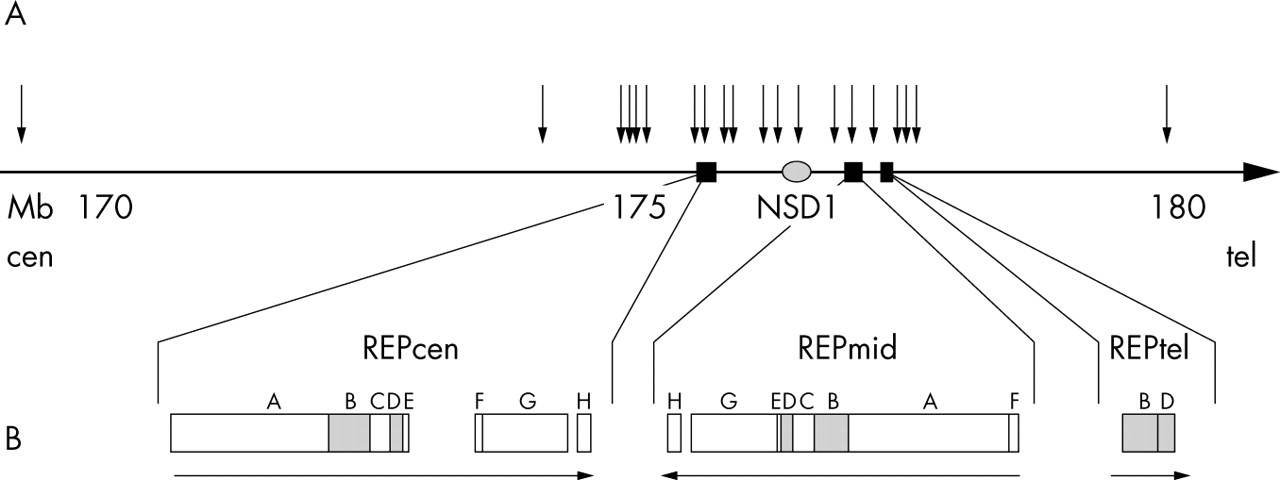
We identified three duplicated areas flanking NSD1 (fig 1). The homology between these regions is very high, generally >98% (table 1). We designated the flanking low copy repeat elements REPcen, REPmid, and REPtel. REPcen is centromeric to NSD1 and consists of eight blocks (REPcenA to REPcenH) ranging in size from 10 to 156 kb (table 1). The total duplicated region is 328 kb, but this is distributed over 400 kb, as there is a 60 kb region between REPcenE and REPcenF and a 12 kb region between REPcenG and REPcenH that is not duplicated in REPmid or REPtel. REPmid is telomeric to NSD1 and is inversely orientated to REPcen. It contains all eight blocks but their order is cen-H-2 kb-GEDCBAF-tel. REPtel is 60 kb telomeric to REPmid and in the same orientation as REPcen. It consists only of blocks B and D (fig 1B).
Sotos microdeletions are variable in size
We analysed 20 microsatellite markers in 33 cases with a microdeletion. Parental samples, if available, were also analysed. These analyses confirmed the presence of a microdeletion in all cases. SOT26 was duplicated in block REPcenH and REPmidH and therefore provided information about centromeric and telomeric breakpoints. The size of the deletion ranged from 482 kb (COG111) in whom NSD1 was the only known gene deleted, to 5 Mb (COG025) in whom 54 known genes were deleted. There were at least eight unique deletion sizes (fig 2). Deletion mapping in four cases, COG343, COG344, COG346, and COG347, suggested the breakpoints may have occurred in REPcen and REPtel giving a deletion size of ∼1.9 Mb. A further 14 cases may also have breakpoints within these repeat elements, but the remaining 15 cases are not consistent with breakpoints in both REPcen and REPtel. The clinical features of the microdeletion cases are shown in table 2.
Sotos microdeletions are primarily generated by interchromosomal rearrangements and are usually paternally derived
We investigated the mechanism generating microdeletions in 11 cases where DNA from grandparents and/or siblings was available. Eight deletions, of varying sizes, arose through interchromosomal rearrangements (COG064, COG111, COG183, COG184, COG344, COG346, COG347, COG512). Two deletions of different sizes arose through intrachromosomal rearrangements (COG231, COG044). One cases was the result of a terminal deletion (COG025) (fig. 3).
We determined the parent of origin of the deletion in 21 cases for whom parental DNA was available. The paternal allele was deleted in 18/21 cases and the maternal allele was deleted in the remaining three cases. The paternally derived deletions included several that were of different sizes and the three maternally derived deletions included the smallest and largest deletion. The bias towards deletion of the paternal allele was statistically significant using the two tailed exact binomial test (p = 0.0015).
DISCUSSION
Identified low copy repeats flanking NSD1 may mediate some, but not all, microdeletions
Through in silico analyses, we identified three low copy repeats (LCRs); one centromeric (REPcen) and two telomeric (REPmid, REPtel) to NSD1. REPcenBD and REPtelBD are ∼50 kb in size, in the same orientation and show >98% homology, and are therefore potential substrates for non-allelic homologous recombination (NAHR), a mechanism implicated in the generation of several microdeletion syndromes.13 Up to 18 of 33 microdeletions may be attributable to NAHR between REPcen and REPtel, resulting in a recurrent ∼1.9 Mb deletion. However, in 14 of these cases, the microsatellite analyses were uninformative at multiple markers, and therefore these deletions may not be of uniform size and some may be attributable to other mechanism(s). Fifteen microdeletions were not consistent with NAHR between REPcen and REPtel, including at least seven distinct deletions. We did not identify other LCRs flanking NSD1, which could mediate these non-recurrent deletions, although it is possible these are present but were not detected by our analyses, as recently demonstrated in non-recurrent deletions in Smith-Magenis syndrome.14 It is also noteworthy that the region encompassing NSD1 has a high density of Alu repeats; 18.8% compared with an average of 10.6% for the human genome.15Alu repeats may act as substrates for homologous recombination, and an increased density of Alu elements is often observed in regions associated with genomic rearrangements.13 Detailed mapping of deletion breakpoints will be required to clarify the mechanisms generating recurrent and non-recurrent Sotos microdeletions.
The paternal allele is preferentially deleted in the majority of cases with NSD1 microdeletions
There was a significant bias towards deletion of the paternally derived allele in our cases with Sotos microdeletions, consistent with previous data from Japanese cases.16 The combined data demonstrate that the paternally derived chromosome was deleted in 36/41 reported cases (p<0.001). It is likely that this bias is, at least in part, attributable to the greatly increased recombination rate in men compared with women at the 5q telomere. In general, the rate of recombination in men is increased at telomeres compared with centromeres, whereas women show a more even recombination rate along the length of a chromosome.17 Other telomeric microdeletions, for example of 4p16.3 in Wolf-Hirschhorn syndrome,18 and of 22q13,19 also show a paternal bias and markedly increased recombination rates in men compared with women, supporting a role for sex dependent recombination rates in the generation of parental bias in some microdeletion syndromes.
Differences in Sotos microdeletion frequency are not due to case ascertainment bias and may reflect differences in genomic architecture
We identified 15 microdeletions and 123 intragenic mutations in 366 cases from the UK who were all fully screened for mutations and deletions of NSD1. These cases were ascertained from across the UK and consisted of a broad range of phenotypes, including individuals with overgrowth but no other features of Sotos syndrome and cases with facial similarity to Sotos syndrome but no overgrowth. There was no obvious bias towards ascertainment of cases with mutations in these analyses. Indeed, as the sensitivity of mutation detection is likely to be less than that of deletions, any bias is likely to be towards identification of cases with microdeletions. Furthermore, all the cases with deletions had been clinically diagnosed with Sotos syndrome prior to the molecular analyses. Our results therefore indicate that ∼10% of Sotos cases in the UK are caused by 5q35 microdeletions. Kurotaki et al reported 49 microdeletions in 95 cases from Japan.11 The identification of such a large number of deletions in Japan, despite the smaller number of cases analysed, suggests there is a genuine difference in microdeletion frequency between the two populations. Our data do not support the hypothesis that this is due to underascertainment of cases with deletions compared with mutations.11 An alternative hypothesis is that differences in genomic architecture in Japanese and non-Japanese populations influences the microdeletion frequency. It is interesting in this regard that only 2/11 cases in our series were generated through intrachromosomal rearrangements, compared with 6/8 Japanese cases.16 Moreover, we identified at least eight distinct deletions, and at least 15/33 deletion cases were not consistent with a recurrent microdeletion, whereas 46/50 Japanese microdeletions were reported to be the same size.11 These data are consistent with possible mechanistic differences in microdeletion generation in Japanese and non-Japanese populations. One possible explanation could be an inversion polymorphism between the inversely orientated LCRs, REPcen and REPmid, being more frequent in Japan, which could predispose to deletions in offspring, as reported in Williams and Angelman syndromes.20,21 However, further analyses of both Japanese and non-Japanese cases with Sotos and microdeletions and their parents will be required to elucidate the processes responsible for the difference in Sotos microdeletion frequency in these populations.
ELECTRONIC DATABASE INFORMATION
The URLs for data presented are as follows.
-
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/OMIM/ (for Sotos syndrome, Wolf-Hirschhorn syndrome, Williams-Beuren syndrome)
-
Primer 3, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
-
RepeatMasker, http://www.repeatmasker.org
-
University of California-Santa Cruz Genome Bioinformatics, http://genome.ucsc.edu/
APPENDIX 1
Primer sequences for 12 new 5q35 microsatellite markers
Size and position of low copy repeat elements flanking NSD1
The clinical features of cases with 5q35 microdeletions

Schematic representation of 10 Mb region surrounding NSD1. (A) Positions of low copy repeat elements REPcen, REPmid, and REPtel, and NSD1. Black arrows represent the microsatellite markers analysed. The names and positions of theses microsatellite markers are shown in fig 2. SOT26 is shown twice as it is duplicated in REPcenH and REPmidH. (B) Enlarged representation demonstrating block composition and orientation.

Results of microsatellite analyses in cases with a 5q35 microdeletion. *Name and position of first base of primer. †The cases are approximately arranged in order of the largest to smallest deletion size. Data were uninformative (U) or unobtainable (N) at some markers in some cases and thus precise deletion size could not always be determined. ‡FGFR4 is an intragenic probe in the SALSA PO26 NSD1 MLPA kit. §D, deleted; shaded box, retained; U, uninformative data; N, no data. ¶COG508 represents deletion in identical twins COG508a and COG508b.

{kind=link}
{kind=link}
{kind=link}
Microsatellite marker analyses in 11 families showing mechanism of generation of 5q35 microdeletions. Haplotypes of marker alleles at 12 microsatellite markers are demonstrated. Filled and open bars next to marker alleles represent the most likely segregation of parental haplotypes above and below deletions. In some cases, other interpretations of the haplotypes are possible but would require additional recombination events. n, no data.
Acknowledgments
K Tatton-Brown is supported by the Birth Defects Foundation and A Irrthum by Tenovus the Cancer Charity. This research was funded by The Child Growth Foundation and the Institute of Cancer Research (UK).
REFERENCES
Footnotes
-
The Childhood Overgrowth Collaboration includes the following contributors: M Addor, A Al Swaid, S Andries, H Archer, A Barnicoat, M Barrow, J Barwell, G Baujat, K Becker, J Berg, B Bernhard, M Bhat, M Bitner, E Blair, A Brady, L Brueton, K Chandler, C Christensen, A Clarke, J Clayton-Smith, T Cole, L Colleaux, A Colley, A Collins, V Cormier-Daire, S Danda, S Davies, R Day, De Roy Magali, N Dennis, A Dobbie, F Elmslie, F Faravelli, H Firth, D Fitzpatrick, N Foulds, J Franklin, A Fryer, S Garcia, C Gardiner, C Garrett, B Gener, R Gibbons, Y Gillerot, D Goudie, A Henderson, J Hirst, S Hodgson, S Holder, T Homfrey, H Hughes, B Kerr, A Kumar, D Kumar, W Lam, N Leonard, J Liebelt, P Lunt, S Lynch, A Magee, S Mansour, M McEntagart, C McKeown, S McKee, K Metcalfe, S Mohammad, A Murray, A Nemeth, S Park, M Patton, E Penny, D Pilz, B Plecko, C Pollitt, S Price, O Quarrell, A Raas-Rothschild, N Rahman, W Raith, J Rankin, L Raymond, W Reardon, E Reid, E Rosser, D Ruddy, H Santos, GB Schaeffer, A Schulze, A Shaw, S Smithson, M Splitt, F Stewart, H Stewart, M Suri, E Sweeney, K Tatton-Brown, I K Temple, E Thompson, M Tischowitz, J Tolmie, S Turkmen, P Turnpenny, Van Maldergem, P Vasudevan, I Vaz, D Waggoner, C Verellen, E Wakeling, D Weaver, K White, L Wilson, R Winter, P Zack, A Zankl.
-
Competing interests: none declared