Article Text

Abstract
Background:BRCA1 is a tumour suppressor with pleiotropic actions. Germline mutations in BRCA1 are responsible for a large proportion of breast–ovarian cancer families. Several missense variants have been identified throughout the gene but because of lack of information about their impact on the function of BRCA1, predictive testing is not always informative. Classification of missense variants into deleterious/high risk or neutral/low clinical significance is essential to identify individuals at risk.
Objective: To investigate a panel of missense variants.
Methods and results: The panel was investigated in a comprehensive framework that included (1) a functional assay based on transcription activation; (2) segregation analysis and a method of using incomplete pedigree data to calculate the odds of causality; (3) a method based on interspecific sequence variation. It was shown that the transcriptional activation assay could be used as a test to characterise mutations in the carboxy-terminus region of BRCA1 encompassing residues 1396–1863. Thirteen missense variants (H1402Y, L1407P, H1421Y, S1512I, M1628T, M1628V, T1685I, G1706A, T1720A, A1752P, G1788V, V1809F, and W1837R) were specifically investigated.
Conclusions: While individual classification schemes for BRCA1 alleles still present limitations, a combination of several methods provides a more powerful way of identifying variants that are causally linked to a high risk of breast and ovarian cancer. The framework presented here brings these variants nearer to clinical applicability.
- DBD, DNA binding domain
- BRCA1
- mutation
- transcription
- tumour suppressor
Statistics from Altmetric.com
Individuals carrying inactivating germline mutations in the breast and ovarian cancer susceptibility gene BRCA1 have an increased risk of developing cancer, making it essential to identify those at risk.1 This task is complicated by the presence of over 1000 different BRCA1 alleles in the population carrying nonsense, missense, frameshift mutations as well as large and small deletions (Breast Cancer Information Core, BIC: http://research.nhgri.nih.gov/bic/). Progress has been made recently in identifying which alleles are likely to be associated with disease. Several lines of evidence derived from population based analysis and functional studies indicate that all mutations leading to premature termination are associated with increased cancer susceptibility.2–4 However, missense mutations still pose an important problem for risk assessment because of their low frequency and, in some cases, ethnic specificity, which make population based studies difficult. Over 300 missense sequence variants have been identified in BRCA1, located throughout the gene (BIC database). In breast–ovarian cancer families in which a missense variant is the only sequence alteration detected, it is difficult to determine whether the variant is causally linked to predisposition or not and so it is uninformative for predictive testing purposes.
Functional studies in which specific activities of the protein or broad biological phenotypes are assayed have contributed to the classification of missense variants.4–11 When integrated with population based studies, functional tests can be a powerful method to help characterise these mutants. BRCA1 is involved in maintaining genomic stability and participates in the DNA damage response, but its biochemical functions have remained elusive.12,13 The BRCA1 protein contains several motifs and structural domains that have been functionally characterised or, in some cases, inferred from sequence analysis and prediction. A zinc binding RING finger,14 which binds the BRCA1 associated RING domain protein BARD1 is present at the N-terminal region (aa 24–64).15 The BRCA1–BARD1 complex behaves as an E3 ubiquitin ligase.15 Cancer associated missense mutations that are located in this region disrupt BRCA1–BARD1 interaction and affect its in vitro ubiquitination activity.16–18 At the C-terminus, two BRCT domains in tandem (BRCT-N: aa 1653–1736; BRCT-C: aa 1760–1855) display a transcription activation function when fused to a heterologous DNA binding domain6,19 and mediate the interaction of BRCA1 with the RNA polymerase II holoenzyme.20 BRCT domains are protein–protein interaction domains found in proteins involved in DNA repair and cell cycle control.21,22 Mutations that result in the truncation or structural alteration of the BRCT segment have been identified in hereditary breast–ovarian cancer families, showing the essential nature of this portion of the gene (BIC database). Importantly, cancer associated missense mutations located at the BRCT domains abolish its transcription activity in an artificial system.4,6,8,19 The strong correlation between cancer association and disruption of a certain biochemical function, even though determined in an artificial system, suggest that specific biochemical tests are powerful tools to characterise these variants.
We and others have characterised several missense variants located at the BRCT domains using a transcription activation assay.4–8,19 While the published data on BRCA1 suggest a (direct or indirect) function in transcription, it is unlikely that BRCA1 represents a bona fide transcriptional activator. Our working model is that the transcription assay is a monitor of the integrity of the C-terminal domain of BRCA1 and therefore can be used to derive functional information. Along those lines, we hypothesised that unclassified missense variants located in regions of BRCA1 that contribute to transcription activation besides the BRCT domains might be amenable to a transcription based classification. In the present study we show that the transcriptional activation assay can be used as a test to characterise mutations in the region encompassing aa 1396–1863 (exons 13 to 24) and we specifically investigated 13 missense variants (H1402Y, L1407P, H1421Y, S1512I, M1628T, M1628V, T1685I, G1706A, T1720A, A1752P, G1788V, V1809F, and W1837R). In addition, we analysed all the mutations using a prediction algorithm based on interspecific sequence variation and Grantham matrices.23 Pedigrees were also analysed for segregation analysis, and posterior probabilities were calculated to determine the odds of causality for each variant. Finally, co-occurrence of the variant with other known deleterious mutations was taken into account. These results were combined with previously published results derived from methods including a prediction algorithm based on general protein structure parameters that evaluates the impact on function for mutations at the BRCT domain,24 and a protease based assay.25 This integrated approach provided us with a cross validated scheme to classify variants as well as to identify the strengths and limitations of current methods.
METHODS
Constructs
Wild type GAL4 DNA binding domain (DBD) fusion construct aa 1560–1863 of human BRCA1 in pGBT9 (Clontech) was previously described.4 The following wild type BRCA1 fragments were amplified by polymerase chain reaction (PCR) using the plasmid pcBRCA1-385 (a gift from Michael Erdos, National Human Genome Research Institute) as a template and the following nucleotide primers: fragment 2–11B (aa 1–323) (ZNF2F, 5′GCGGAATTCATGGATTTATCTGCTCTT 3′; BRAL, 5′ATAGTCGACTTCCAGCCCATCTGTTATGT 3′), 13–21 (aa 1396–1778) (UX13, 5′CGGAATTCCAGAGGGATACCATGCAA 3′; LX21DM, 5′GCGGTCGACATCTGTGGGCATGTT 3′), 14–24 (aa 1455–1863) (UX14, 5′CGGAATTCACTTCACAGAAAAGTAGT 3′; 24ENDT, 5′GCGGATCCTCAGTAGTGGCTGTGGGGGAT 3′), 13–24 (aa 1396–1863) (UX13, 5′CGGAATTCCAGAGGGATACCATGCAA 3′; 24ENDT), 12–24 (aa 1366–1863) (UX12, 5′CGGAATTCGGTGAAGCAGCATCTGGG 3′; 24ENDT). PCR products were gel purified and digested with EcoRI (2–11B only), EcoRI and SalI (13–21 only), or EcoRI and BamHI, and fragments were ligated in frame to the GAL4 DBD of the yeast expression vector pGBT9 similarly digested. Construct 11B24 (aa 302–1863) was made by digesting pGBT9 16–24 with EcoRI and NcoI and isolating a 6.1 kb fragment containing the vector and the end of BRCA1 coding region. Subsequently, pcBRCA1–385 was similarly digested and a 4.0 kb fragment (EcoRI-NcoI) was isolated and ligated to the previous pGBT9 fragment, generating a pGBT9 (aa 302–1863). To obtain the full length, a PCR fragment corresponding to exons 2–11B (aa 1–323) was digested with EcoRI, and a 1 kb fragment was isolated and ligated to a similarly digested pGBT9 11B24.
The yeast expressing vector pLex9 carrying a wild type BRCA1 sequence (aa 1396–1863) fused in frame to the LexA DNA binding domain (DBD) was used as wild type control and as a backbone to introduce the mutations described in table 1 by site directed mutagenesis using the following methods. Mutations S1613G, A1708E, M1775R, and Y1853X were subcloned from previously described constructs.5,6 Mutation H1402Y was introduced by direct PCR using primers 24ENDT and H1402Y-U (5′ GGAATTCCAGAGGGATACCATGCAATATAACC 3′). Mutations L1407P, A1752P, and G1706A were introduced by the Quickchange (Stratagene) method according to the manufacturer’s instruction. The following primers containing the alteration of interest were used for PCR involving the wild type constructs produced in a methylation competent bacterial strain, and amplification was carried out using Pfu polymerase. L1407P (L1407P-U, 5′ CTGATCAAGCCCCAGCAGG 3′; L1407P-L, 5′ CCTGCTGGGGCTTGATCAG 3′); G1706A (G1706AF, 5′ GACACTGAAATATTTTCTAGCAATTGCCGGCGGAAAATGG 3′; G1706AR’, 5′ CCATTTTCCGCCGGCAATTGCTAGAAAATATTTCAGTGTC 3′); A1752P (A1752PF, 5′ CCAAGGTCCAAAGCGACCTCGAGAATCCCAGGAC 3′; A752PR, 5′ GTCCTGGGATTCTCGAGGTCGCTTTGGACCTTGG 3′). Dpn1 was subsequently added to digest the parental plasmid leaving only cDNAs with the introduced mutations to be transformed into bacteria.
Comprehensive analysis of BRCA1 variants
Mutations H1421Y, S1512I, P1614S, M1628T, M1628V, T1685I, T1700A, T1720A, G1788V, V1809F, and W1837R were introduced using splicing by overlapping extension PCR.26 The first round of PCR was undertaken using the following primers: H1421Y 3′ region (H1421Y-U2, 5′ GTTAGAACAGTATGGGAGCCAGCCTTCT; 24ENDT); H1421Y 5′ region (H1421Y-L2, 5′AGAAGGCTGGCTCCCATACTGTTCTAAC; UX13); S1512I 3′ region (S1512I-U, 5′ GGTACATGCACATATGCTCTGG-3′; 24ENDT); S1512I 5′ region (S1512I-L, 5′ CCAGAGCATATGTGCATGTACC 3′; UX13); P1614S 3′ region (P1614S-U, 5′ GCAGAATCTGCCCAGAGTTCAGCTGCTG 3′; 24ENDT); P1614S 5′ region (P1614S-L, 5′ CAGCAGCTGAACTCTGGGCAGATTCTGC 3′; UX13); M1628T 3′ region (M1628TF, 5′ GCCGGCTATAATGCAACGGAAGAAAGTGTGAGCAGG 3′; 24ENDT); M1628T 5′ (M128TR′, 5′ CCTGCTCACACTTTCTTCCGTTGCATTATAGCCGGC 3′; UX13); M1628V 3′ region (M1628VF, 5′ GCCGGCTATAATGCAGTGGAAGAAAGTGTGAGCAGG 3′; 24ENDT) M1628V 5′ region (M1628VR′, 5′ CCTGCTCACACTTTCTTCCACTGCATTATAGCCGGC 3′; UX13); T1685I; T1700A 3′ region (T1700A-U, 5′ GTTTGTGTGTGAACGGGCACTGAAATAT 3′; 24ENDT); T1700A 5′ region (T1700A-L, 5′ ATATTTCAGTGCCCGTTCACACACAAAC 3′; UX13); T1720A 3′ region (T1720A-U, 5′ GCTATTTCTGGGTGGCCCAGTCTATTAA 3′; 24ENDT); T1720A 5′ region (T1720A-L, 5′ TTAATAGACTGGGCCACCCAGAAATAGC 3′; UX13); G1788V 3′ region (G1788V-F, 5′ GCTGTGTTGCTTCTGTGG 3′; 24ENDT); G1788V 5′ region (G1788V-R, 5′ CCACAGAAGCAACACACAG 3′; UX13); V1809F 3′ region (V1809F-F, 5′ CCAATTGTGTTTGTGCAG 3′; 24ENDT) V1809F 5′ region (V1809F-R, 5′ GGCTGCACAAACACAATTGG 3′; UX13); W1837R 5′ region (W1837R-F, 5′ GGTGACCCGAGAGCGGGTGTT 3′; 24ENDT); W1837R 3′ region (W1837R-R, 5′ CCAACACCCGCTCTCGGG 3′; UX13). For each mutation, both products (5′ and 3′ regions) were combined and used as a template for a final round of PCR using 24ENDT and UX13 primers. The final PCR products were cloned into pCR-Script Amp SK(+) vector (Stratagene). The inserts were then isolated by cutting with BamH1 and EcoR1 and ligated to pLex9 or pGBT9 vectors. All mutations were confirmed by sequencing. To obtain GAL4-DBD fusions in a mammalian expression vector, pGTB9 constructs were digested with HindIII and BamH1, a 1.8 kb band was isolated and ligated into equally digested pCDNA3.
Transcription assay in yeast
Two Saccharomyces cerevisiae strains were used: EGY48 [MATa, ura3, trp1, his3, 6 lexA operator-LEU2] and SFY526 [MATa, ura 3-52, his3-200, ade 2-101, lys s-801, trp 1-901, leu 2-3, 112, canr, gal 4-542, gal 80-538, URA3::GAL1-lacZ].27 SFY526 cells contain a lacZ reporter gene under the control of GAL1 UAS, which is recognised by GAL4 DNA binding domain (DBD). EGY48 cells were transformed with plasmid reporters pSH18–34, pJK103, or pRB1840 which contain a lacZ gene under the control of eight, two, and one LexA operators, respectively.27,28 Competent yeast cells were obtained using the yeast transformation system based on lithium acetate (Clontech) and cells were transformed according to the manufacturer’s instructions. At least three individual EGY48 or SFY526 clones for each variant were tested for liquid β-galactosidase assays using ONPG,29 and the assays were carried out in triplicate. The β-galactosidase activity was noted as a comparison to wild type BRCA1 and S1613G (positive controls) or to A1708E, M1775R, and Y1853X (negative controls). Western blot analysis was carried out as previously described.4
Transcription assay in mammalian cells
We used pG5Luc, which contains a firefly luciferase gene under the control of five GAL4 binding sites, as a reporter for the assay. Transfections were normalised with an internal control phGR-TK (Promega), which contains a Renilla luciferase gene under a constitutive TK basal promoter using a dual luciferase system. Human 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% calf serum and plated in 24-well plates the day before transfection. Transfections were done in triplicate using Fugene 6 (Roche) and harvested 24 hours post-transfection. Cells were lysed in RIPA buffer (150 mM NaCl, 10 mM Tris-HCl pH 7.4, 5 mM EDTA, 0.1% sodium dodecyl sulphate, 1% Triton X-100, 0.1% sodium deoxycholate). The blots were incubated with α-GAL4 DBD monoclonal antibody (Clontech). Lysates were cleared and samples were separated on 10% SDS-PAGE; equal amounts of protein were loaded for every sample. Gels were electroblotted on a wet apparatus to a polyvinylidene difluoride (PVDF) membrane and probed with an α-LexA DBD monoclonal antibody (Clontech).
Our laboratory has completed the analysis of 27% of the existing unclassified variants (32/117) in the C-terminus of BRCA1 (residues 1560–1863). To validate the assay we have used (a) all the unambiguously classified missense variants, and (b) all other variants for which there are strong (but not definitive) clinical data. Using the set of variants in (a), the assay correctly classified the four variants that can be classified unambiguously as deleterious (A1708E, R1699W, M1775R) or benign (S1613G) based on clinical data. Using the set of variants in (b), the assay correctly classified all nine other variants with strong supporting clinical evidence for classification. These results suggest a high sensitivity and specificity for this assay.
RESULTS
Frequency in control populations
Six variants (G1706A, A1708E, A1752P, M1775R, G1788V, and W1837R) were assessed using PCR/LDR followed by assessment in a DNA microarray30 in 500 healthy women without cancer of varying ages (18–40 years) and ethnic groups. All were at a frequency of 0/500. The remaining variants were assessed by denaturing gradient gel electrophoresis (DGGE) with the following frequencies: H1402Y (0/500), L1407P (0/500), H1421Y (0/500), S1512I (6/500), S1613G (260/500), M1628V (0/500), M1628T (0/500), T1685I (0/500), W1837R (0/500), and Y1853X (0/500). Variants T1720A and V1809F were not assessed.
Activation of transcription
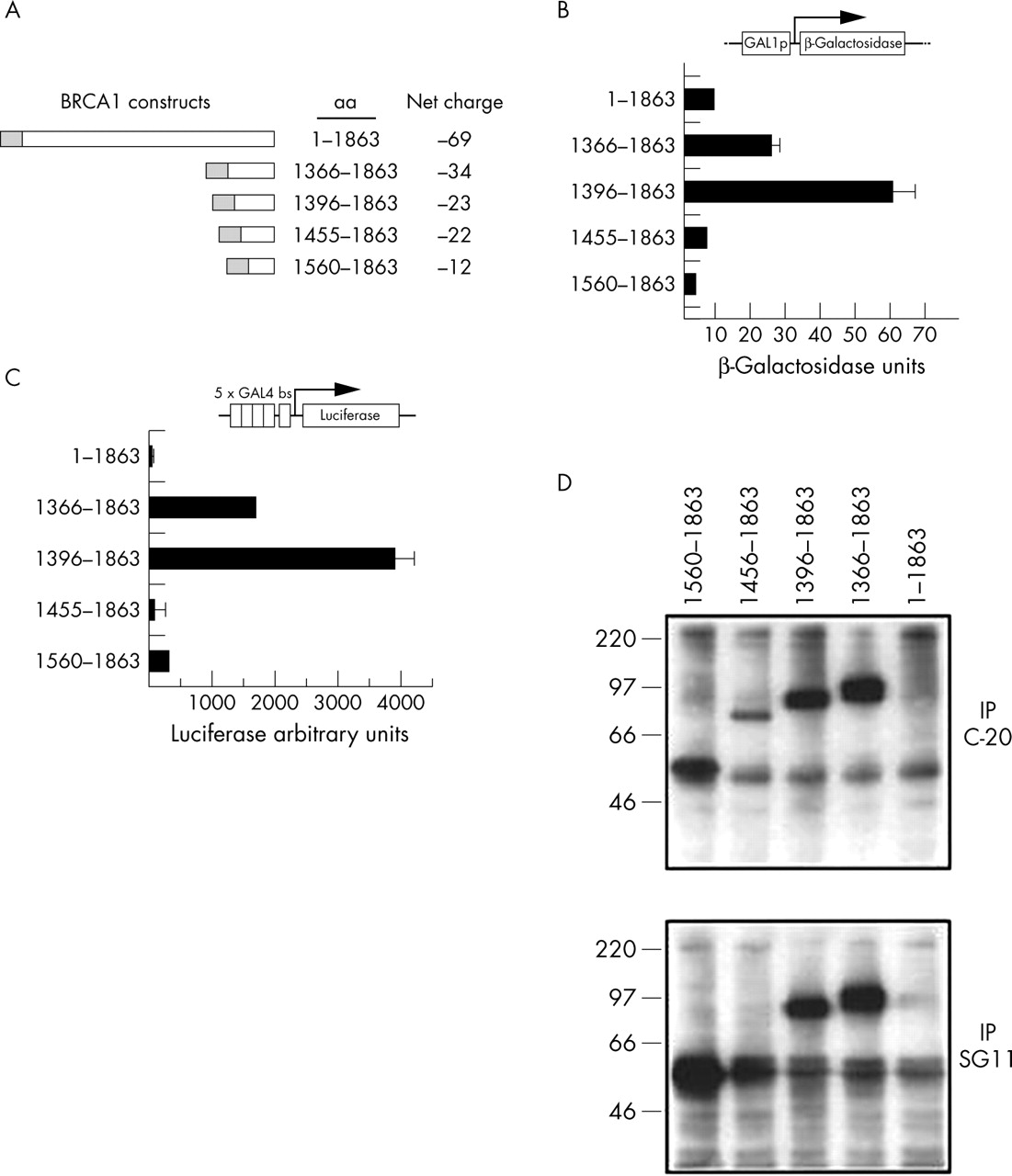
Regions of BRCA1 that contribute to its transcription activation function besides aa 1560–1863 might also be used in a transcription based functional assays to classify missense variants. In order to determine their contribution we quantitatively assessed activation of a β-galactosidase reporter gene under the control of a GAL4 responsive promoter by a series of GAL4DBD:BRCA1 fusion constructs in yeast (fig 1A). These constructs were designed to test systematically the contribution of exons 13 to 24 to activation.

Regions of BRCA1 that contribute to transcription activation. (A) Fusion constructs of GAL4 (yeast and mammalian cells) or LexA (yeast) DNA binding domain (grey boxes) to wild type BRCA1 used in this study. Net charges of BRCA1 sequences are indicated. (B) Presence of activating and inhibitory regions in BRCA1. Yeast cells (SFY526) were transformed with the designated constructs, and transcriptional activity was measured by the lacZ reporter gene. Cells were grown in liquid culture and β-galactosidase activity was assayed. Each bar corresponds to three independent transformants that were assayed in triplicate. (C) Activation of transcription in mammalian cells: 293T cells were co-transfected with the GAL4 DBD fusion constructs and a luciferase reporter gene under the control of GAL4 responsive elements. Four independent experiments done in duplicate are shown. Standard deviation was less than 15% in all cases (not shown). (D) Immunoprecipitations with α-BRCA1 C-terminal polyclonal antibody (C-20, directed against aa 1843–1862; top panel) or monoclonal antibody (SG11, directed against aa 1846–1863; bottom panel), blotted against α-GAL4DBD monoclonal antibody.
While a construct including aa 1455–1863 did not show activity that was significantly higher than aa 1560–1863, a construct containing aa 1396–1863 had an activity that was markedly higher (fig 1B). The results in a mammalian expression system confirmed those obtained in yeast, with construct aa 1396–1863 showing the highest activity (fig 1C). In this case, however, the contribution of aa 1455–1559 is less clear, but this may reflect instability of the aa 1455–1863 construct in mammalian cells. We can detect expression of this construct when using a polyclonal antibody (fig 1D, second lane), but it is not recognised by a monoclonal antibody raised against a similar epitope (compare second lane in fig 1D, top and bottom panel). We were unable to obtain significant levels of expression of the GAL4DBD full length protein in 293T cells, even after several attempts.
BRCT-N domain alone is not capable of activating transcription
Previous experiments indicated that the BRCT-C repeat alone could activate transcription when fused to a heterologous DBD. Although activity was abolished by disruption of the BRCT-C repeat, it was still possible that the first domain could show residual activity. To test for this, we made a GAL4 fusion construct aa 1396–1778, which includes BRCT-N (aa 1653–1736) but not BRCT-C (aa 1760–1855). Sequences contained in aa 1396–1559 were defined as the strongest auxiliary activating region in BRCA1 (see above), providing the most favourable context to demonstrate any residual activity. This construct did not show any significant activation (not shown), indicating that this region can collaborate to augment activity but cannot act alone. Although there is a correlation of negative charge and transcription activation, charge does not seem to be the only determinant (fig 1). The fragment aa 1560–1863 is less negatively charged than the aa 1396–1778 fragment, yet the former is able to activate transcription while the latter is not. In conclusion, sequences outside the BRCT domains confer higher activity to BRCA1 but cannot act alone. The construct containing exons 13–24 (aa 1396–1863) showed the highest transcription activation (15-fold activation ability over full length BRCA1) being the most sensitive to detect differences in transcription activation and it was therefore chosen as the backbone in which to introduce the sequence variants for the transcription activation assay.
Functional analysis of missense variants
The location of the 13 missense variants studied as well as the negative and positive controls are indicated by arrows in fig 2A. Seven variants lie in the BRCT domains. Six of the variants lie upstream of the BRCT domains, three of which lie within the putative coiled coil domain (fig 2A). Three known BRCT deleterious/high risk variants—A1708E, M1775R, and Y1853X—were used as negative (that is, loss of function) controls, and S1613G (a common neutral polymorphism) and wild type BRCA1 (aa 1396–1863) were employed as positive controls. Results show that both in yeast and mammalian cells the three negative controls located in the BRCT domain lose most of transcription activation function consistent with a loss of function mutation, whereas the positive control S1613G had an activity equal to or higher than the wild type, as observed previously5,6 (fig 2B and 2C). Seven unclassified missense variants in the BRCT domains (T1685I, G1706A, T1720A, A1752P, G1788V, V1809F, and W1837R) were then tested. Variants T1685I, A1752P, G1788V, V1809F, and W1837R showed greatly decreased transcription activation levels (at least <50%) both in yeast and mammalian cells, comparable with the known mutant controls and consistent with their classification as deleterious/high risk variants (fig 2B and 2C).

Functional analysis of missense variants in BRCA1. (A) Location of variants (blue), negative and positive controls (red and green, respectively). Grey boxes, BRCT domains; DBD, GAL4 DNA binding domain; blue box, putative coiled-coil domain. (B) Quantitative assay in yeast. (C) Quantitative assays in mammalian cells. (D) Protein levels were determined by western blots in yeast (upper panel) and mammalian cells (lower panel).
Variants G1706A and T1720A showed slightly reduced transcription activation levels in yeast cells, at 64% and 74%, respectively, of the wild type control. Interestingly, whereas T1720A had activity comparable to wild type in mammalian cells, G1706A showed a markedly reduced activity. The intermediate results for these mutants suggest that they may represent moderate rather than high risk variants.
Six additional variants (H1402Y, L1407P, H1421Y, S1512I, M1628T, and M1628V) in locations outside the BRCT domains were also investigated for their effect on transcription. Three of the variants were located in a region in which a putative coiled coil domain has been predicted to form (fig 2A).31 Variant L1407P showed significantly reduced transcription activation levels consistent with a high risk mutation (fig 2B, 2C). Variants H1402Y, H1421Y, and S1512I showed transcription activation levels equal to or higher than wild type BRCA1, suggesting that they do not represent high risk variants and are likely to have low clinical significance.
In yeast cells, protein levels were slightly variable in three independent clones. Most variants displayed levels comparable to wild type, with the exception of Y1853X and T1685I, which showed markedly reduced levels suggesting that protein instability might be the underlying cause of loss of function. In mammalian cells, some variants (S1512I, V1809F, and W1837R) had markedly reduced levels. However, no loss of function variant showed consistently reduced levels in yeast and mammalian cells, suggesting that even when expressed at higher levels they were unable to activate transcription (fig 2D).
Pedigree analysis
In order to obtain additional information to classify the missense variants we applied a recently developed full likelihood method for the evaluation of causality from family data.32 For the analysis of co-segregation we assumed an allele frequency of the variant of 0.0001 and a penetrance model with separate age specific risks of breast and ovarian cancer for BRCA1 based on meta-analysis estimates.33 We obtained six pedigrees for five variants (M1628T, G1706A, T1720A, V1809F, and W1837R) (fig 3). For M1628T we obtained odds against causality of 10.4:1, consistent with the data obtained in the functional assay. For G1706A we obtained odds against causality of 1.3:1. This rather uninformative result reflects the fact that this large pedigree with multiple cases typed had one case diagnosed at age 53 that did not carry the variant. This result may also reflect the fact that G1706A may be a moderate rather than a high risk variant. For variant V1809F the odds in favour of causality were 7.3:1, consistent with the functional test, suggesting that it is a high risk variant. Two pedigrees were analysed for the T1720A variant generating combined odds against causality of 355:1. This result is also consistent with the functional data and suggests that T1720A represents a neutral/low clinical significance variant. For variant W1837R we obtained odds of 4:1 against causality, which contradicts our functional data.

{kind=link}
{kind=link}
{kind=link}
Pedigrees of missense variants in BRCA1. The presence (M) or absence (WT) of the variants in the germ line of tested individuals is indicated. Affected individuals are denoted by a black circle or square; site of tumour and age of diagnosis are also indicated.
Analysis of interspecific sequence variation
In order to determine further the likelihood that a particular variant may or may not represent a high risk variant we also analysed the amino acid substitution using a modified Grantham matrix, adapted for BRCA1.23 These results are shown in table 1. The classification based on interspecific sequence variation confirms our choice of controls, with S1613G being classified as neutral and M1775R, A1708E, and Y1853X being classified as deleterious. It also confirms our functional results for H1402Y, L1407P, T1685I, G1706A, G1788V, and W1837R. Variant M1628V, however, was classified as a neutral/low risk variant in contradiction of our functional results. The remaining variants could not be classified by this method.
Co-occurrence with deleterious mutations
Homozygous disruption of Brca1 in mouse resulted in embryonic lethality (reviewed by Brodie and Deng44). In addition, there is a deficit from expected numbers of BRCA1 homozygotes and compound heterozygotes for deleterious mutations among individuals with the founder mutations 185delAG and 5382insC.45 This led to the notion that if an unknown variant co-occurs with a known deleterious mutation it is unlikely that this variant is a high risk one. Co-occurrence data relative to a set of 40 000 individuals (kindly provided Amie Deffenbaugh, Myriad Genetics Laboratories Inc) are listed in table 1. Variants H1402Y, S1512, and M1628T co-occur with a deleterious mutation and are therefore unlikely to represent high risk variants, a result supported by the functional assays.
DISCUSSION
In order to provide a more informative risk assessment for individuals carrying a mutation in BRCA1 we used several approaches, including association studies and segregation analysis. However, the clinical relevance of missense variants has been particularly elusive because of their low frequency, making it difficult to conduct meaningful population based studies. Several lines of investigation must be considered in the classification of a BRCA1 variant into deleterious/high risk or neutral/low clinical significance.46 The occurrence of the variant in high risk individuals (affected by breast or ovarian cancer and with a family history of breast or ovarian cancer) compared with controls can provide clues as to its status, but the frequencies of variants differ considerably between ethnic groups.36 Segregation of the variant in the family members affected by the disease may be a strong indicator that a variant is deleterious except if it is in linkage disequilibrium with another mutation, as observed in some ethnic groups.47 Additional approaches have relied on sequence comparisons23,48–50 or on functional tests which include specific assays such as transcription activation, or broader phenotypes such as the generation of a yeast small colony phenotype or induction of apoptosis in cultured cells.7–9,11
One method of classification of a BRCA1 variant is the transcription functional assay. We have previously shown that alleles containing neutral polymorphisms retained wild type activity in transcription.5 Several lines of evidence have pointed to a physiological role of BRCA1 in transcription, although its exact biochemical function is unclear.20,51–56 However, regardless of whether or not BRCA1 acts as a transcription activator in vivo, we have proposed that a transcription assay using a heterologous DNA binding domain fusion and a reporter gene serves as a monitor of the integrity of the C-terminal region of BRCA1. Because this region is essential for the tumour suppressive function of BRCA1, the transcription assay is able to generate information about the impact of missense changes. Previously, this assay was only applicable to variants in the BRCT domain (exons 16–24). Here we show that regions adjacent to the BRCT domains contribute to full activity in transcription, allowing us to extended our analysis to encompass exons 13–24. Once it was verified that the extended assay assigned positive and negative controls correctly, seven missense variants in the BRCT domain and six variants outside the BRCT domain were tested (fig 2). The variants were chosen because they were identified as the sole BRCA1 alteration in individuals considered to be at high risk for breast or ovarian cancer (S1512I, M1628V, M1628T, T1685I, G1706A, T1720A, A1752P, G1788V, V1809F, and W1837R) or were located at or in close proximity to the putative coiled-coil domain (H1402Y, L1407P, and H1421Y).
Seven of the variants caused a dramatic loss of the transcription activation function (L1407P, M1628V, T1685I, A1752P, G1788V, V1809F, and W1837R), similar to known high risk mutation controls, suggesting that they may constitute deleterious/high risk variants (fig 2B and 2C; table 1). Four of the variants (H1402Y, H1421Y, S1512I, and M1628T) showed transcription activation similar to or greater than the wild type BRCA1, suggesting they are probably neutral/low clinical significance variants. The two remaining variants had intermediate results. G1706A showed a slightly reduced activity in yeast but a markedly reduced activity in mammalian cells. Variant T1720A had a slightly reduced activity in yeast and but activity comparable to wild type BRCA1 in mammalian cells. Based on the results obtained we would tentatively classify T1720A as a neutral/low clinical significance variant. At this point we cannot classify variant G1706A.
We then carried out an analysis using interspecific variation, pedigree analysis, and co-occurrence data (table 1). Of the seven variants for which the method based on interspecific variation was able to reach a conclusion, the data confirmed the classification based on our functional tests in six cases (L1407P, H1402Y, T1685I, G1706A, G1788V, and W1837R) but presented a contradiction for variant M1628V. The pedigree analysis confirmed our results for three the four variants analysed. Results from pedigree analysis for W1837R contradicted all the other methods, although the odds against causality were rather small. Interestingly, results for G1706A suggested again that the available information is not enough to classify it. While no conclusion can be drawn for variants that have very low frequency and are not found to co-occur with a deleterious mutation (for example, L1407P and H1421Y), co-occurrence data indicated that H1402Y, S1512, and M1628T do not represent high risk variants, confirming the functional assay results.
In order to further cross validate our analysis we compared it with results from three published methods to classify variants in the BRCT domain (table 1). The first method is based on the fact that variants that cause conformation changes are more likely to be prone to proteolytic degradation.25 For all five of the variants analysed by this method (T1720A, A1752P, G1788V, V1809F, and W1837R), protease sensitivity correlated with abrogation of transcriptional activation. We also compared our data with results derived from a method based on protein structure parameters to predict the outcome of different variants of BRCA1.24 For the seven variants for which there is a prediction, six (T1685I, G1706A, T1720A, A1752P, G1788V, and W1837R) confirmed the results obtained. In fact, the G1706A variant was considered not explained by the algorithm because the qualitative yeast data used to test for G1706A indicated wild type function while the algorithm predicted a functional impact. It is possible that G1706A may represent a moderate/low risk variant and our current methods are not yet powerful enough to recognise this. Variant V1809F was contradictory; however, given that fact that pedigree analysis, transcriptional activation, and protease sensitivity indicate a high risk variant, our conclusion is that, although the change is a conservative one, the side chain size threshold in the algorithm needs to be refined. In summary, we have classified six missense variants (L1407P, M1628V, T1685I, A1752P, G1788V, and V1809F) as probable deleterious/high risk variants and the remainder as probable neutral/low clinical significance variants (H1402Y, H1421Y, S1512I, M1628T, and T1720A), with two variants (G1706A and W1837R) left unclassified.
Previously, all the known deleterious missense changes were in the RING domain or BRCT repeats. Although further work is needed to classify variants L1407P and M1628V unambiguously, our results provide evidence that other regions or motifs are likely to harbour high risk missense substitutions. In particular, classification of L1407P as a high risk variant suggests an important function for the putative coiled coil motif as previously suggested.31,57 The 4-3 spacing of hydrophobic residues in the coiled coil is clearly evolutionarily conserved through the puffer fish BRCA1 sequence (Tetraodon, accession AY428536).
Conclusions
While individual classification schemes for BRCA1 alleles still present limitations and no single method can reliably be used alone, a combination of several methods may provide a more powerful way of identifying variants that are causally linked to a high risk of breast and ovarian cancer.58 The framework presented here pushes our understanding of these variants further towards clinical applicability in the near future.
Acknowledgments
We thank Qun Wang for excellent technical assistance and Barbara Pasini (University of Torino) for additional pedigree information. This work was supported by AMDeC Foundation of New York City (FB), US Army awards DAMD17-00-1-0478 (CMP), DAMD17-99-1-9389 (ANAM), NIH CA92309 (ANAM), CAM 8.1/0018.1/03 (TC), Italian Association and Foundation for Cancer Research (PR), and the Italian Ministry of Health (Ricerca Finalizzata 2002; PR). VD is a postdoctoral fellow of the New York State Board of Health and Education. KConFaB (a list of members can be found at http://www.kconfab.org/organisation/members.asp) has been funded by the Kathleen Cuningham Foundation, National Breast Cancer Foundation, National Health and Medical Research Council (NHMRC), Cancer Council of Victoria, Cancer Council of South Australia, Queensland Cancer Fund, Cancer Council of New South Wales, Cancer Foundation of Western Australian, and Cancer Council of Tasmania.
REFERENCES
Supplementary materials
In our paper "Classification of BRCA1 variants of unknown clinical significance" we reported that BRCA1 variant M1628V presented markedly reduced activity which suggested that it represented a deleterious variant. During the course of additional experiments (Carvalho and Monteiro, unpublished data) we noticed that constructs containing the M1628V variant described in our paper also included a 3' mutation leading to a truncated protein that was overlooked. We have now generated correct constructs for this variant and performed extensive testing. Contrary to what was reported this variant displayed activity comparable to the wild type BRCA1 suggesting that it corresponds to a neutral variant. We regret this error and apologize for any confusion or inconvenience it may have caused.
Footnotes
-
↵* These authors contributed equally to this work
-
Conflicts of interest: none declared