Article Text

Abstract
Introduction: Interpretation of results from mutation screening of tumour suppressor genes known to harbour high risk susceptibility mutations, such as APC, BRCA1, BRCA2, MLH1, MSH2, TP53, and PTEN, is becoming an increasingly important part of clinical practice. Interpretation of truncating mutations, gene rearrangements, and obvious splice junction mutations, is generally straightforward. However, classification of missense variants often presents a difficult problem. From a series of 20 000 full sequence tests of BRCA1 carried out at Myriad Genetic Laboratories, a total of 314 different missense changes and eight in-frame deletions were observed. Before this study, only 21 of these missense changes were classified as deleterious or suspected deleterious and 14 as neutral or of little clinical significance.
Methods: We have used a combination of a multiple sequence alignment of orthologous BRCA1 sequences and a measure of the chemical difference between the amino acids present at individual residues in the sequence alignment to classify missense variants and in-frame deletions detected during mutation screening of BRCA1.
Results: In the present analysis we were able to classify an additional 50 missense variants and two in-frame deletions as probably deleterious and 92 missense variants as probably neutral. Thus we have tentatively classified about 50% of the unclassified missense variants observed during clinical testing of BRCA1.
Discussion: An internal test of the analysis is consistent with our classification of the variants designated probably deleterious; however, we must stress that this classification is tentative and does not have sufficient independent confirmation to serve as a clinically applicable stand alone method.
Statistics from Altmetric.com
Interpretation of results from mutation screening of tumour suppressor genes known to harbour high risk susceptibility mutations, such as APC, BRCA1, BRCA2, MLH1, MSH2, TP53, and PTEN, is becoming an increasingly important part of clinical practice. In most cases, interpretation of frameshift and nonsense mutations, large gene rearrangements, and obvious splice junction mutations, is quite straightforward. However, classification of missense variants observed in these genes often presents a difficult problem.
Classifying missense variants is difficult because clinical testing is generally performed on small families or isolated individuals as opposed to the large pedigrees frequently used for linkage analysis, coupled with the fact that the individual missense variants in question are usually very rare. As a result, these variants cannot be analysed using the typical human genetics approaches of linkage analysis or allelic association. Another difficulty in classifying missense variants is that, for the data to be clinically useful, they often must be generated in the context of a cost effective test that can be run at high throughput with reasonable turnaround time. We can imagine biochemical assays, either in vitro or in organisms with quick phenotype readouts such as bacteria or yeast, that would meet the requirements. In fact, several such assays have been developed1–5; unfortunately, they lack generality as they are specific to individual domains or individual functions of the proteins. A family of approaches that circumvents both of these difficulties emerges from detailed sequence analysis of the genes in question, either in the context of cross species multiple sequence alignment, crystallographic structure, or both. Once the groundwork required to institute alignment based methods as a clinical test is in place, they have the advantage of being inexpensive and essentially instantaneous; the main barrier to their use is in satisfying the requirement that the resultant classification be sufficiently supported to provide a basis for clinical decision making.
The theme of using analysis of multiple protein sequence alignments in a medical genetic context has been developing for some time. Recognition that such alignments provide a window on the evolutionary history of the proteins involved6 led to the deduction that the positions in such alignments at which only a single amino acid is observed are conserved because amino acid substitutions at these conserved positions often adversely effect protein function, and also to the deduction that variable positions are variable because at least some substitutions at these positions are not deleterious.7 This logical connection provides a framework through which an evolutionary biology perspective can be applied to multiple sequence alignments, enabling their application to human disease genetics. Indeed, there are recent demonstrations that the long term sequence evolution/conservation pattern revealed by multiple sequence alignment of orthologues of a human disease susceptibility gene across an appropriately diverse set of organisms does have some power to distinguish human disease susceptibility missense changes from missense changes of little clinical significance.8–11
Myriad Genetic Laboratories, Inc has been performing a gene resequencing test for susceptibility to familial breast and ovarian cancer, BRACAnalysis®, since 1996. By late 2002, Myriad Genetic Laboratories had completed sequencing BRCA1 and BRCA2 from more than 20 000 individuals, most with a personal or a family history of breast or ovarian cancer. A wide variety of sequence alterations was observed in the two genes (a public database of sequence variants observed in these genes is maintained at http://research.nhgri.nih.gov/bic/). The total number of unique BRCA1 and BRCA2 variants seen in this time period was 891 and 1277, respectively. The number of clearly deleterious mutations or mutations strongly suspected to be deleterious found by resequencing (so not including the results of genotypic analysis for large genomic rearrangement mutations) was 407 and 411, respectively. Three hundred and fourteen of the remaining BRCA1 variants were missense changes and eight were in-frame deletions. Only 21 of these missense changes were classified as deleterious or suspected deleterious and 14 as neutral or of little clinical significance. The analysis reported here attempts to classify the remaining 279 missense changes and eight in-frame deletions.
MATERIALS AND METHODS
Identification of puffer fish BRCA1
tBLASTn searches12 of the puffer fish Fugu rubripes genomic sequence13 with human and Xenopus BRCA1 RING finger sequences, BARD1 RING finger sequences, BRCA1 BRCT repeats, and BARD1 BRCT repeats revealed two Fugu sequence scaffolds (scaffolds 1510 and 3928, but note that scaffold numbers may have changed in more recent assemblies) containing both a RING finger sequence and BRCT repeats. Multi-exon gene models encompassing these RING finger and BRCT repeats were created, translated, and searched against GenBank. The result was that the scaffold 1510 RING and BRCT sequences had their strongest hits against BRCA1s from multiple species, while the scaffold 3928 RING and BRCT sequences had their strongest hits against BARD1s from multiple species. We concluded that the Fugu genome encodes a single BRCA1 orthologue residing on scaffold 1510 and BARD1 orthologue on scaffold 3928.
Using a combination of splice junction prediction and peptide sequence similarity between the known tetrapod BRCA1s and predicted open reading frames at the Fugu BRCA1 locus, we created tentative gene models for the Fugu BRCA1 RING finger and BRCT domains. In addition, examination of the Fugu genomic sequence between these well conserved domains revealed a long open reading frame with some sequence similarity to the large central exon (exon 11) of human BRCA1. However, we did not find it possible to create, with a high level of confidence, a complete BRCA1 gene model from Fugu genomic sequence alone.
As a partial genome sequence of the green spotted puffer fish Tetraodon nigriviridis was also available, we carried out the same analysis on the Tetraodon sequence, with a very similar result. To obtain a complete, correct puffer fish BRCA1 coding sequence, PCR based methods were used to amplify and sequence from Tetraodon cDNA.
Sequencing of Tetraodon BRCA1
One green spotted puffer fish was dissected, and total RNA was prepared from the brain and internal organs using TRI Reagent (Molecular Research Center). Total RNA was reverse transcribed using Superscript II (Invitrogen) and the tailed random primer 3’N10 ([biotin]-CTCCACCTCGAGAACGCATTTGTCCN10). Heteroduplex cDNA was converted to double stranded cDNA by the RNaseH/DNA polI method. After end polishing, the cDNA was made ready for 5′ RACE by ligation of the double stranded, non-phosphorylated oligo 5 tag (5′ [Cy5]-CAGGAATTCAGCACATACTCATTGTTCAGAA annealed to TTCTGAACAATGAGTATGTGCTGAATTCCTX (where X is a non-nucleotide residue such as a C5-spacer)).
The vast majority of Tetraodon BRCA1 was PCR amplified from cDNA using forward and reverse primers based on confidently predicted exonic sequences and a 20:1 mix of Taq Platinum and Platinum Pfx polymerases (Invitrogen). Primers were selected so that predicted product lengths would be between 1 kb and 2 kb, and so that the products would overlap to allow complete sequencing. In cases where several bands were observed on agarose gels, individual bands were plugged with a Pasteur pipette and re-amplified. PCR reactions were digested with shrimp alkaline phosphatase and Exonuclease I and then sequenced using Big-Dye dye-terminator chemistry (ABI), using either the original PCR primers or internal primers derived from predicted or observed exon sequences. Samples were subject to electrophoresis on MegaBACE 1000 capillary electrophoresis instruments (Molecular Dynamics/Amersham Life Sciences) and sequences were called with software developed at Myriad Genetics, Inc (MGI) and some hand editing. As RT-PCR was directly from fish cDNA and no cloning was involved in the process, we observed a number of heterozygous positions in the cDNA sequence. Where missense causing heterozygous positions were observed, the base call observed from a corresponding genomic sequence chromatogram (publicly available sequence chromatograms from shotgun cloning and sequencing of the Tetraodon genome), was used to pick the allele of our reference sequence. These positions were: L140F, E197K, H413N, T507A, T575M, H689Y, K838I, V932L, M956I, M956T, S1037N, and L1199M (where 1 = the initiator methionine).
5′ RACE was used to PCR amplify across the start codon and into the 5′ UTR. RACE was carried out using a nested PCR strategy where the first amplicon used the forward primer 5-ampA (CAGGAATTCAGCACATACTCA) and an outside gene specific reverse primer, and the nested amplicon used the forward primer 5-ampB (TTCAGCACATACTCATTGTTCA) and an inside reverse primer. The primary RACE PCR was carried out as a cycle titration, and products were subject to electrophoresis on an agarose gel. Size fractions ranging from 250 bp to 1000 bp were gel purified from the cycle number that was just sufficient to generate a visible smear over this size range (in this case, 25 cycles). After these size fractions were re-amplified and run out on agarose gels, clear bands were gel purified and sequenced with the nested reverse sequencing primer plus upstream primers (based on the sequencing results). As an in-frame stop codon was found upstream of the predicted start codon, and both were readily observed in corresponding Fugu genomic sequence, assignment of the translation initiator was clear.
Creation and analysis of the multiple sequence alignment
The multiple sequence alignment was made with the multiple sequence alignment program T-Coffee14 using BLAST-P and CLUSTAL-W to generate the underlying high-scoring segment pairs. GenBank accession numbers for BRCA1 protein sequences used in the alignment were as follows: Human, NP_009225; Chimpanzee, AAG43492; Mouse, AAD00168; Dog, AAC48663; Chicken, AAK83825; Xenopus, AAL13037; Tetraodon, AY428536 (this last is a nucleotide sequence accession). A parsimony based method was used to calculate the minimum number of missense substitutions required to create the observed alignment, taking into account the underlying phylogenetic tree.15 Because we are interested in human disease genetics, our subsequent analyses only considered sequence variation at positions in the alignment where the human sequence has a residue. On the other hand, absence of an amino acid in the alignment of a non-human BRCA1 at a position where the human sequence does have a residue was considered a sequence variation.
Analysis of sequence invariance in the alignments
We have considered three models for calculating, from the multiple sequence alignments and underlying cladograms, the number of positions in the alignment that remain invariant due to common ancestry and strong sequence conservation as against those that are invariant due to common ancestry even though the position is probably under weak functional constraint. All three models follow from Fitch’s development of the covarion hypothesis.15–17 For each model, L is the number of positions in the alignment where human BRCA1 has a residue. Depending on the model, there are three classes of positions: invariant positions with substitution rate λ1 = 0 (Class 1), slowly substituting positions with substitution rate λ2 (Class 2), and rapidly substituting positions with substitution rate λ3 (Class 3).
Model 1
The probability of missense substitutions and indels does not depend on position in the sequence; rather, it is modelled with a single substitution rate constant resulting in a single Poisson distribution of positions with 0, 1, 2,…k substitutions. Only one variable is fitted, the substitution rate constant λ3.
Model 2
The alignment is treated as if it consisted of two groups of positions, one group of invariant (Class 1) positions and one group of variable (Class 3) positions. Amino acid substitution at the variable positions is modelled with a single substitution rate constant. The expected distribution of positions with 0, 1, 2,…k substitutions is the sum of a single Poisson distribution over the Class 3 positions and the number of Class 1 positions, which have no substitutions. Two variables are fitted, the number of Class 1 positions, L1, and the substitution rate constant that applies to the Class 3 positions, λ3.
Model 3
The alignment is treated as if it consisted of two groups of positions, one group of slowly substituting (Class 2) positions and one group of rapidly substituting (Class 3) positions. Amino acid substitution is modelled with two substitution rate constants, one for the Class 2 and one for the Class 3 positions. The expected distribution of positions with 0, 1, 2,…k substitutions is the sum of a Poisson distribution over the Class 2 positions and a second Poisson distribution over the Class 3 positions. Three variables are fitted: the number of Class 2 positions, L2, the substitution rate constant that applies to the Class 2 positions, λ2, and the substitution rate constant that applies to the Class 3 positions, λ3.
The expected number of invariant positions, N0, for all three models can be calculated by using the following expression:

where Di,n is the degree of sequence divergence among Class i positions that has accumulated within each clade n with corresponding substitution rate λi (independent of the other clades) and m is the number of non-human sequences in the alignment. For model 1, L1 = L2 = 0, and L3 = L; for model 2, L2 = 0, and L1+L3 = L; finally, for model 3, L1 = 0, and L2+L3 = L.
The expected number of positions with 1,2,…k substitutions requires more complicated calculations that take into account the possible location of the substitutions in the tree. For example, in the case of a position with one substitution, we have to consider all possible locations of the substitution among m clades:

Determining in this way all expected numbers Nk, we find such values of Li and λi that provide the best fit of Nk to the actual numbers listed in table 1.
Number of expected and observed positions with different numbers of missense changes
Estimations of the expected number of Ashkenazi homozygotes and compound heterozygotes
There are two BRCA1 founder mutations in the Ashkenazi population, 185delAG and 5382insC. No Ashkenazis tested at Myriad have been found to be either homozygotes or compound heterozygotes for these mutations. Should we expect to have seen any? If q1 and q2 are the population allele frequencies of two independent alleles of BRCA1, and N1 and N2 are the number of carriers of those two alleles observed in our test series, then, under the conservative assumption that the trait is dominant, the equilibrium number of homozygotes and compound heterozygotes of these two alleles expected in our test series, Nexp, is

Description of the BRCA1 component of BRACAnalysis®
All analyses of BRCA1 and the two prevalent mutations in Ashkenazi individuals were performed by direct gene sequencing as previously described.18 For a test to be performed, all test request forms must be signed by the ordering health care provider indicating that “informed consent has been signed and is on file”. Patient samples were each assigned a unique bar code for robotic specimen tracking. Most samples were received as 7 ml of anticoagulated blood, from which DNA was extracted and purified from leucocytes isolated from each sample. Aliquots of patient DNA were each subjected to polymerase chain reaction (PCR) amplification (35 reactions for BRCA1 or two reactions for analysis of the two Ashkenazi founder mutations analysed in Ashkenazi samples). The amplified products were each directly sequenced in the forward and reverse directions using fluorescent dye-labelled sequencing primers. Chromatographic tracings of each amplicon were analysed by Myriad Genetics’ sequence analysis software followed by visual inspection and confirmation, assisted by comparison of the proband sequence to a consensus wild-type sequence constructed for each amplicon. Multiple amplicons were reviewed in parallel by trained staff in such a way that the identification of one mutation did not influence the review of the remaining sequence data. Each genetic variant (exclusive of non-reportable polymorphisms) was independently confirmed by repeated analysis including PCR amplification of the indicated gene regions and sequence determination.
All mutations and genetic variants were named according to the convention of Beaudet and Tsui,19 and all of them have been submitted to the BIC database, http://research.nhgri.nih.gov/bic/. Nucleotide numbering starts at the first transcribed base of BRCA1 according to GenBank entry U14680. (Under this convention, the two mutations commonly referred to as “185delAG” and “5382insC” are named 187delAG and 5385insC, respectively. However, in this paper we will refer to those two variants by the more commonly used “185delAG” and “5382insC”).
RESULTS
Sequence conservation in BRCA1
The most common uses of multiple sequence alignments are to determine the evolutionary relationships between species (phylogenetic reconstruction) and to identify DNA, RNA, or protein functional motifs. Here we have another purpose: to use sequence conservation observed within a protein multiple sequence alignment as a tool to help categorise potentially disease related missense changes and in-frame deletions in a disease susceptibility gene. To do this, we must first construct an appropriately informative multiple sequence alignment of homologous coding sequences of the gene of interest. In preparing such an alignment, two potentially conflicting considerations need to be balanced: (a) maximisation of the amount of sequence diversity sampled in the alignment, and (b) solid evidence that all of the sequences sampled in the alignment are functionally equivalent.
Because we are interested here in human disease genetics, consideration (a) need only be subject to the limitation that when the species from which sequences are being used are viewed on a cladogram, the number of sequences sampled from each clade not containing human sequences should be equal. Consideration (b) is, however, more problematic. It is clear, for instance, that during functional diversification of the members of a gene family, the diverging paralogues are subject to an unusually high rate of sequence evolution, which can include missense substitutions at functionally important residues.20 We might hypothesise that, along with restricted temporal and spatial patterns of expression, such missense changes help create functional specificity for diverging gene paralogues. Because of this, the biological phenomenon of functional diversification among paralogues makes it best to restrict disease genetics analyses to sequence alignments of true orthologues. It should also be recognised that second site suppression occurring during protein sequence evolution, either in cis or in trans, will create a false impression of sequence variability at specific positions in a multiple sequence alignment and thereby degrade the ability to distinguish between conserved and variable sites in the alignments.
To a certain extent, complete model organism genome sequences provided by various offshoots of the human genome project are simplifying the process of assembling the multiple sequence alignments required for the analysis of any specific gene. For instance, keeping in mind that a predicted gene model from a genomic sequence contig does not entirely substitute for a sequenced cDNA, it is much easier to clone and sequence highly diverged orthologues by the combination of sequence search and RT-PCR than by hybridisation cloning, protein interaction cloning, or PCR cloning using degenerate primers. Moreover, orthologues and paralogues can be distinguished with much more confidence, if the complete genome sequences can be queried, rather than a limited number of cDNA or EST sequences.
BRCA1 is a member of a very small gene family characterised by the presence of a RING finger in the amino half of the protein and a pair of BRCT repeats at the C-terminus.21 In vertebrates studied to date, the only two members of this gene family are BRCA1 and BARD1. Global analysis of the Fugu genomic sequence13 revealed the presence of 254 RING finger proteins, but did not specifically identify either BRCA1 or BARD1 orthologues. tBLASTn searches of the Fugu genomic sequence with human and Xenopus BRCA1 and BARD1 sequences readily revealed that both are present in the Fugu rubripes genome as single copy genes. A similar strategy applied to the Tetraodon nigriviridis genome sequence www.genoscope.cns.fr/externe/tetraodon/Ressource.html yielded a similar result. However, we were not confidently able to construct a complete BRCA1 gene model directly from genomic DNA sequence of either fish. Due to issues of mRNA availability, we chose to PCR amplify (using a combination of RT-PCR and 5′-RACE) and sequence BRCA1 from Tetraodon rather than Fugu. Like mammalian BRCA1, we found Tetraodon BRCA1 to have a non-coding first exon and a very long central exon. The gene encodes a protein of 1267 amino acids, with a RING finger near its N-terminus, a large central exon corresponding to human exon 11, a clear coiled coil domain near the BRCT repeats, and a pair of BRCT repeats at its C-terminus. Even though the protein is structurally quite similar to human BRCA1, its sequence is quite diverged, sharing only 19% overall sequence identity with the human protein (table 2).
Pairwise percent sequence identities among BRCA1 sequences
To make an analysis of BRCA1 in which all segments of the protein are equally represented, we chose to use only full length coding sequences. Available sequences were from human, chimpanzee, mouse, rat, dog, chicken, Xenopus, and Tetraodon. Following the rule of using an equal number of sequences per clade, we arbitrarily chose to use the mouse rather than the rat sequence. The protein multiple sequence alignment was made with the multiple sequence alignment program T-Coffee,14 with some hand optimisation (DF and SVT). Pairwise percent sequence identities from the alignment are given in table 2, a cladogram showing the relationships among the species sampled is given in fig 1, and the alignment itself is given in fig 2.
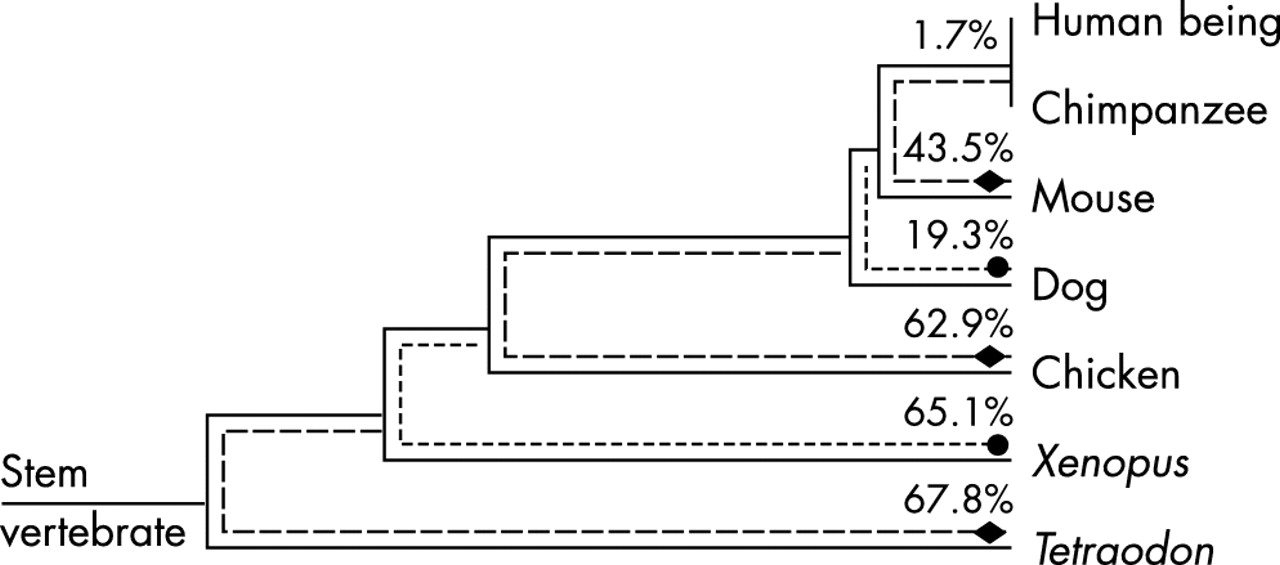
Cladogram: stem lengths are proportional to the amount of time since the indicated most recent common ancestor or the amount of time between indicated common ancestors.22–25 The evolutionary paths over which we are calculating minimum % sequence changes are indicated by dotted lines ending in a diamond or circle (except for human/chimp, for which there is no space). The minimum calculated percentage changes are given along each such path.
{kind=link}
{kind=link}
BRCA1 multiple sequence alignment. Green cells denote positions at which one or more missense variants have been classified as neutral or of little clinical significance. Red cells denote positions in the alignment at which one or more missense variants have been classified as deleterious; the subset of these where the classification is due to interfering with a splice junction is denoted with an “Sp” just above the residue. Pink cells denote positions in the alignment at which we find one or more currently unclassified missense variants likely to be deleterious. Cells with a green outline denote positions at which we find one or more currently unclassified missense variants that are probably neutral or of little clinical significance; at the subset of these where the human sequence variant matches the canonical residue in another species, that residue also has a green outline. One position (R1699) has both a known deleterious variant and a variant that we find to be probably deleterious. One position (I15) has both a variant that we find to be probably deleterious and one that we find to be probably neutral or of little clinical significance. Positions of the conserved segments described in table 3 are annotated by heavy grey bars.
With only 55.8% amino acid sequence identity between human and mouse and only 74.6% identity between human and dog (table 2), BRCA1 is among the most weakly conserved protein coding genes in the mammalian genome. At 1863 amino acids (human), it is also one of the larger proteins in the genome. Even cursory examination of the full alignment reveals that some positions are invariant across all seven species represented. Given the evolutionary breadth of the alignment, one would normally assume that all of these invariant positions are under strong functional constraint. On the other hand, one might argue that, given that a large fraction of this protein must consist of relatively weakly conserved amino acid positions and that it should take a number of years for the time dependent processes of nucleotide substitution and amino acid substitution to result in an amino acid substitution at every such position in the alignment, there may remain some weakly conserved positions in the alignment that are invariant.
To begin our analysis of BRCA1, we calculated the minimum number of missense substitutions or indels at each of the 1863 positions in the multiple sequence alignment where the human sequence has a residue required to create the observed alignment from a hypothetical most recent common ancestor sequence. Using the minimum number of required substitutions (as described in Materials and methods), we found that this could be accomplished with 4820 changes, and that 137 positions appeared invariant, 230 positions required one change, 422 positions required two changes, 624 positions required three changes, 376 positions required four changes, and 74 positions required five changes.
If, during the evolution of BRCA1, missense changes happened with the same probability across the entire protein coding sequence, this would mean that the entire sequence is under relatively weak function constraint and that all of the observed invariant positions are actually weakly conserved. The alternative possibility is that there are positions within BRCA1 where the probability of a missense change really is much lower than the average for the whole protein. To address this, we compared the number of invariant positions actually observed with an estimate of the number of invariant positions that we should have observed under the assumption that missense changes happened with the same probability all over the gene (model 1). For this model, the estimated average amino acid substitution frequency across the whole sequence alignment is 3.9 per position and the expected number of positions at which no changes should be observed is 37 (table 1). This estimate is much lower than the 137 invariant positions that we observed (binomial test p value = 4×10−39) and leads us to reject the hypothesis that a large fraction of invariant positions are actually weakly conserved positions.
It is quite clear that there are some positions in the BRCA1 sequence at which the probability of missense variations is much lower than at others, even though some subset of the invariant positions may actually be weakly conserved invariant. Addressing this issue, we developed model 2, which assumes that some positions of the gene are conservative and do not accept any changes. The best fit to the actual BRCA1 data was observed for a substitution frequency of 4.5 per position and 133 conservative positions. This model gives a better estimate for the number of invariant positions, 153, but significantly underestimates the number of positions with a single substitution: 160, as against 230 observed from BRCA1 data (table 1).
Since many more positions have lower variability than one would expect due to weakly conserved invariance alone, we recalculated the expected number of invariant positions under the more realistic assumption that there are well conserved segments within BRCA1 in which the probability of a missense substitution is much lower than in the rest of the protein. In model 3, we assumed that there are two classes of positions with low and high substitution rates, respectively. The best fit to the BRCA1 data was observed for 281 conservative positions with a substitution frequency of 0.8 per position, which is ≈6 times lower than in the non-conserved positions, 4.9 substitutions per position (table 1). Because the successive models that we have used differ from each other by one degree of freedom, the relative goodness of fit between the successive models is approximated well by Δχ2 between those models, with a p value drawn from the χ2 distribution at 1 degree of freedom. Importantly, χ2 for model 3 is just 2.3. Since the 1 degree of freedom p value for this is >0.05, no model that has to fit more parameters (more than the three required for model 3) can give a significantly better fit. Accordingly, we shall conditionally use two conclusions derived from model 3: (a) >90% of the invariant positions observed in the alignment are actually under strong functional constraint, and (b) a small but appreciable fraction of invariant positions, ≈9%, are likely to be weakly conserved positions.
As has been reported by others, the invariant positions are not randomly distributed over BRCA1 but tend to form clusters.21,27,30 If missense variations were distributed randomly, the expected median distance between 137 invariant positions would be 9.6 amino acids. In reality it is only 2.0 amino acids, a clear sign of the strong clusters. In addition, the positions in the alignment that are adjacent to invariant positions have considerably less amino acid variability than the average over the entire BRCA1 alignment, even after excluding adjacent positions that are themselves invariant (data not shown). Upon inspection of the alignment, we find eight clearly conserved segments; their positions, lengths, and degrees of conservation are summarised in table 3, and their positions are annotated on fig 2. Of these eight segments, four have been well described: these are the amino terminal RING finger domain,26 the two carboxy terminal BRCT repeats,29 and a coiled coil domain which is located 200–300 residues proximal to the first BRCT repeat.28 In an analysis of BRCA1 based on a multiple sequence alignment in which the most diverged sequence was from chicken,27 the four well described conserved segments plus seven additional conserved motifs were described. Of these seven more recently described motifs, three are absent from our alignment, while the remaining four (labelled motifs 3, 4, 6, and 7 in table 3 and on fig 2) are all conserved through Tetraodon. Interestingly, the purported nuclear localisation signals31,32 are not well conserved.
Clearly conserved segments of BRCA1
Classification of missense variants observed during clinical testing
Having shown that most of the invariant positions in BRCA1 are not weakly conserved invariant, we analysed how missense changes that have been observed during clinical testing correlate with conservation of amino acids among orthologous BRCA1 genes. Our analysis included three steps:
-
Analysis of the limited number of missense variants in BRCA1 that have already been classified,
-
Identification of unclassified variants in BRCA1 that are probably deleterious,
-
Identification of currently unclassified variants in BRCA1 that are probably neutral or of little clinical significance.
In each of these steps, we considered two independent criteria: sequence conservation (or lack of it) at the point in the multiple sequence alignment to which any given missense variant maps, and physicochemical similarity between the variant amino acid and the residues in the alignment at the position to which the given variant maps.
It is well established that the chemical difference between amino acids strongly correlates with the effect of missense changes on phenotype. In the analysis presented below, we have chosen to use the Grantham chemical difference matrix, which is based on a multivariate combination of residue side chain composition, polarity, and volume.33 The advantages of this chemical difference matrix over more commonly used synonymous substitution matrices, such as the PAM or BLOSUM series, have been discussed recently.9 The values in the Grantham chemical difference matrix vary substantially; closely similar pairs of amino acids have scores in the range of 5 to 60, whereas strongly dissimilar pairs have scores of above 100 (the highest score, Cys↔Trp, is 215). The average chemical difference between amino acids wherein the substitution confers a known phenotype (calculated using the data from the Human Genome Mutation Database) is 93.4, the average chemical difference between amino acids based on codon frequencies (which would correspond to the model of random missense changes without evolutionary pressure) is 78.3, and the average chemical difference for polymorphic changes and interspecific variations for several genes is close to 60.9
Of 314 distinct missense changes observed in BRCA1 in the course of 20 000 full sequence tests conducted at Myriad Genetic Laboratories, 21 have been classified as deleterious and 14 as neutral or of little clinical significance (table 4). Most of the deleterious variants were classified by a combination of segregation analysis and functional assay, while most of the neutral variants were classified by a combination of association study and segregation analysis26,34–40 (and Myriad Genetic Laboratories internal studies: see table 4). For the 21 deleterious missense changes, the average chemical difference score is 122.0. However, five of these missense changes were classified because they fall within the splice donor consensus at the end of an exon and have either been shown to interfere with splicing or are strongly predicted to do so. Excluding these five variants from the analysis, the remaining 16 deleterious missense changes have an average chemical difference score of 133.3, and all 16 of them fall at residues which are invariant in the BRCA1 multiple sequence alignment. In contrast, the 14 missense changes that have been classified as neutral or of little clinical significance have an average chemical difference score of 64.8; all 14 happened at positions where at least one interspecific variation was observed and 12 happened at positions where three or more variations were observed. The two neutral missense variants observed at positions with fewer than three substitutions in the sequence alignment are Y856H and P871L. Two missense substitutions are observed at residue 856 and one of them, in Xenopus, is to His. At residue 871, human seems to be the odd species out; all of the others have Leu at this position. The probability that all of the non-splice junction deleterious missense changes would fall at invariant residues is 3×10−19 (binomial test), whereas the probability for 12 or more of the neutral changes to fall at positions where three or more variations were observed is ≈0.02 (binomial test).
Previously classified variants in BRCA1
From our analysis of known deleterious missense substitutions in BRCA1 and missense substitutions that are known to be neutral or of little clinical significance, we have thus established two strong differential correlations. The first is that the position of deleterious missense substitutions, within the multiple sequence alignment, is strongly biased towards invariant residues, whereas the position of neutral substitutions is biased towards positions where multiple substitutions have been accepted during the evolution of this protein. The second is that deleterious missense substitutions tend to have relatively high chemical difference index scores, whereas neutral substitutions tend to have relatively low chemical difference index scores. Similar observations have recently been made using an independent set of human disease susceptibility genes.9 Therefore, we should be able to use these parameters to help determine the status of the 279 missense changes and eight in-frame deletions at 255 different positions, from the 20 000 test BRCA1 data set, that are currently unclassified.
Tentative identification of deleterious missense substitutions
Because our data set is almost entirely derived from analysis of individuals who have either a personal or family history of breast or ovarian cancer, it should be enriched with deleterious mutations in BRCA1. Thus, due to the correlations between deleterious mutations, missense changes at invariant positions, and missense changes with high chemical difference scores, we expect that missense changes at invariant positions will be enriched in our data set and that our data set should be relatively rich in missense changes with high chemical difference scores. Indeed, after excluding the missense changes that have already been classified and the positions at which they occurred, we find that 14.3% of missense and in-frame deletions happened at the remaining invariant positions, which constitute only 6.7% of the whole protein length. The probability for this (or a greater) level of bias towards missense changes at invariant positions is only 1×10−4 (binomial test). In addition, the average chemical difference score for the unclassified missense changes is 77.1, which is about what might be expected for random missense variants but significantly higher than that expected for polymorphic changes or interspecific variation.
We can select two groups of missense variants that are likely to be deleterious substitutions. The first is a set of 40 missense changes that have occurred at invariant positions, and one in-frame deletion that involved an invariant position (table 5). The average chemical difference score for these 40 missense variants is 92.0; this is higher than either the average score for polymorphic changes and interspecific variation or the average score for the set of unclassified missense changes from which they were drawn (a p value of 0.02 was obtained in simulations), and is almost exactly equal to the average for deleterious missense variants from the Human Genome Mutation Database. Thus we are quite confident that the group is highly enriched for deleterious variants. However, the issue remains as to whether all of these variants are deleterious. After the first step of classification of likely deleterious substitutions and in-frame deletions, we were left with observations of unclassified variants at 224 out of the 1863 positions in human BRCA1. Combining these data with our earlier result that we observed 137 invariant positions in the alignment but expected about 12 of these positions to be weakly conserved invariant rather than invariant because of strong functional constraint, we would expect 12×{224/(1863−137)}≈1.6 of these unclassified missense changes and in-frame deletions to fall at a position that is weakly conserved invariant. This gives us one estimate, (41−1.6)/41≈96% of the fraction that are probably deleterious; however, a definite answer as to which specific variants are not deleterious cannot be extracted from the existing data. It would seem sensible that those variants that are not from one of the clusters of conserved positions and those variants with particularly low chemical difference scores are somewhat less likely than the others to be deleterious.
Unclassified variants in BRCA1 that we find likely to be deleterious
As shown in table 1, one, two, and sometimes even three missense substitutions are observed at 55% of the positions within the evolutionarily conserved segments of BRCA1. Inspection of the multiple sequence alignment reveals that, within the conserved segments, positions at which only conservative substitutions are observed cluster with the positions at which no substitutions have been observed. Therefore non-conservative substitutions observed in patients at positions located within conserved segments of the protein and at which only a small number of uniformly conservative substitutions have been observed are likely to be deleterious. Accordingly, we have constructed a criterion to select these missense substitutions from the data set. The criterion has two components. (1) In the multiple sequence alignment, at the positions in question, the highest pairwise chemical difference score ⩽61 (on inspection of the Grantham matrix, this is a good estimate for the upper limit of what might be recognised as a conservative substitution). (2) The chemical difference score for the human missense mutation ⩾3× the highest pairwise chemical difference score in the alignment at that position. When this selection criterion was applied to the database of missense changes, ten more missense variants that are likely to be deleterious were found (table 5). We have also included one in-frame deletion (V1688del) in this list because only a very conservative substitution, V→I, has been observed at that position. All of these changes mapped within one of the conserved segments of BRCA1 are listed in table 3. Nine of these eleven changes are fewer than three positions away from an invariant position (p value = 8×10−7, binomial test). The average chemical difference for the 10 missense changes that we selected through this criterion is 96.7—just above the average for deleterious missense changes from the Human Genome Mutation Database.
It might be argued that inclusion of Xenopus and Tetraodon sequences in our alignment can be misleading because these species are so distantly related to humans, and that the alignment containing just mammalian and chicken sequences is sufficiently informative to identify deleterious missense substitutions. This is an easily testable hypothesis. There are 260 positions in the alignment, constituting 14% of the protein, that are invariant from human to chicken, but substituted in Xenopus, Tetraodon, or both. Thirty missense substitutions and one in-frame deletion (11% of the unclassified variants that we are considering) were observed at one or another of these positions. The average chemical difference for these 30 missense substitutions is 69.3. Thus it appears that the ascertainment criteria for testing do not result in enrichment for mutations at these positions and that the average chemical difference for these substitutions is below that expected for random missense substitutions. Thus we reject the hypothesis and instead argue that the Xenopus and Tetraodon sequences are adding valuable information, from the point of view of identifying high risk missense substitutions in BRCA1, to the alignment. We also argue that in a BRCA1 alignment that captures considerably less time depth, enough weakly conserved invariant positions will remain in the alignment to seriously compromise a method of identifying deleterious missense substitutions based on the preceding logic. For example, from model 3, we can estimate that of the 260 positions that are “merely” invariant from Human to chicken, 69 (27%) are at Class 2, highly functionally constrained positions while the rest are weakly conserved invariant positions. This would lead us to predict that only ≈8 of the 31 missense substitutions or in-frame deletions observed at one of these positions are actually high-risk.
Tentative identification of missense substitutions that are neutral or of little clinical significance
We can also select two groups of missense variants that are likely to be neutral or to have little clinical significance. Missense changes that lead to the consensus BRCA1 sequence of another species are relatively likely to be neutral, especially if the species is closely related to Humans. In addition to six known polymorphisms, 56 unclassified missense variants fall into this category: four between Human and chimpanzee, 15 between Human and mouse, seven between Human and dog, 14 between Human and chicken, 12 between Human and Xenopus, and four between Human and Tetraodon. These are presented in table 6. The average chemical difference for the 56 missense changes with unspecified status which lead to the consensus BRCA1 sequence of another species is 62.7—almost exactly the average for the known neutral changes. This is significantly lower than the average over the 279 missense changes with unspecified status (a p value = 0.003 was obtained in simulations) and in accord with our hypothesis that these missense variants are neutral.
Unclassified variants in BRCA1 that we find likely to be neutral or of little clinical significance
A second set of probable neutral missense variants was selected by a criterion analogous to that used to pick the second set of probable deleterious variants. In contrast to the observation that pairwise chemical differences between amino acids found at conserved positions in the alignment tend to be quite low, pairwise chemical differences between amino acids found at unconserved positions in the alignment will sometimes be quite high. Therefore conservative substitutions found in patients at positions in the alignment where high pairwise chemical difference scores are observed are likely to be neutral or of little clinical significance. Accordingly, we have constructed a two component criterion to select these missense substitutions from the data set: (a) the chemical difference score for the human missense substitution is <61, and (b) the chemical difference score for the human missense substitution should be less than one third of the highest chemical difference score of an interspecific genetic variation at that position. We have found 36 additional missense variants that meet this criterion (table 6). The average chemical difference of these 36 changes is 25.1, which is significantly lower than what we and others have observed for neutral changes in other genes.9 This is not surprising because the criterion used to identify these changes has a systematic bias towards low chemical difference scores.
An internal test for consistency of the classification of missense variants
Are there any other BRCA1 genetics data available with which we can make an independent test of our classification? Several lines of evidence lead to the conclusion that homozygosity or compound heterozygosity for high risk mutations in BRCA1 confers an embryonic lethal phenotype. The first hint came from mouse knockout studies which repeatedly showed that Brca1 null mice had severe developmental defects appearing between days 6 and 9 of embryonic development and leading to embryonic death.41–44 More recently, a targeted mutation directed at the second BRCT repeat, which intentionally leaves most of the mouse Brca1 protein intact, was also shown to confer a homozygous lethal phenotype.45 As these knockouts were created using several different mutations and were constructed on or moved into mice with more than one genetic background, the phenotype is clearly somewhat mutation-independent and strain-independent. In a completely independent animal model, antisense mediated depletion of Xenopus laevis BRCA1 in early stage Xenopus embryos caused severe developmental abnormalities that lead to death of the embryos.21 On the other hand, there are clearly instances where a homozygous frameshift in Brca1 does not uniformly confer an embryonic lethal phenotype. In the best explored instance,46 the phenotype conferred upon mice that were homozygous for a Brca1 exon 11 frameshift varied from embryonic lethal to viable but with clear developmental abnormalities and very high risk of a variety of tumour types.
While it must be acknowledged that one human BRCA1 homozygote has been reported,47 the validity of that report has been strongly questioned on methodological grounds.48 Analysis of data from the Myriad Genetic Laboratories BRACAnalysis® database now supports absence of BRCA1 homozygotes and compound heterozygotes from the testing population. As is clear from equation 3, to calculate the expected number of BRCA1 homozygotes and compound heterozygotes, one has to know the allele frequencies of these mutations in the general population. Accurate estimates of these frequencies are not yet available; however, the frequencies are expected to be so low that even our sample of 20 000 individuals is unlikely to produce interesting results. However, in the Ashkenazi Jewish population, there are two founder frameshift mutations in BRCA1: 185delAG and 5382insC. Taken together, the allele frequency of these two founder mutations in Ashkenazis is higher than the summed allele frequency of high risk BRCA1 mutations in the general white population. The best estimates for the allele frequencies of these two mutations among young cancer free Ashkenazis in the United States and Canada are 0.0053 and 0.0014, respectively.49,50 As a component of BRACAnalysis®, Myriad Genetic Laboratories offers a genotyping test for these two mutations, as well as the Ashkenazi BRCA2 founder mutation 6174delT, called Multisite3. At the time that 20 000 full sequence tests had been completed, 6895 Multisite3 tests had also been completed: in that sample set, 745 individuals were found to carry 185delAG and 222 to carry 5382insC. From the allele frequencies and the observed number of carriers, we would expect from equation 3 to have seen 6.5 homozygotes or compound heterozygotes; in fact, we have seen none (p = 0.002, binomial test). Thus the absence of BRCA1 homozygotes or compound heterozygotes from the Multisite3 data set supports the notion that the phenotype conferred by this genotype prevents carriers from becoming part of the BRACAnalysis® test population.
Because, aside from Multisite3, BRACAnalysis® is a full resequencing test, we know for the individuals tested both whether or not they carried any unclassified missense variant and whether or not they carried any clearly deleterious mutation of the types readily found by the test. Since inheritance of deleterious mutations in both copies of the BRCA1 gene is probably lethal, the most likely way for a single individual to carry two deleterious mutations would be for a second deleterious mutation to occur in a copy of a BRCA1 gene that already had one, which should be a rare event. Therefore, missense changes suspected to be deleterious should only extremely infrequently be observed with known deleterious mutations.
In 20 000 tests performed by Myriad Genetic Laboratories, clearly deleterious BRCA1 mutations were found in 1765 individuals (8.8%). None of them also carried a second clearly deleterious mutation. Among the 774 carriers of 274 unspecified changes (excluding five which are in strong disequilibrium with clearly deleterious mutations) only 41 also carried a clearly deleterious mutation. (p value = 2×10−4, assuming an expected frequency of co-occurrence of 0.088). This clearly indicates that some of the unspecified changes are deleterious. There were a total of 123 carriers of the 50 missense variants and two in-frame deletions that we propose are very likely to be deleterious on the basis of this analysis. None of them carried a second clearly deleterious mutation (p = 1×10−5). Co-occurrence of the changes suspected to be deleterious with each other has also never been observed. In contrast, among the 291 carriers of the 92 missense variants that we propose are likely to be neutral, (after excluding two such variants that were in strong disequilibrium with clearly deleterious variants) there were 18 carriers with an additional clearly deleterious mutation, which is insignificantly different from the frequency of deleterious mutations in the tested population. We cannot be sure that all of the previously unclassified missense changes that we propose as likely to be deleterious are actually deleterious. Nor can we be sure that all of the previously unclassified missense changes that we propose as likely to be neutral are actually neutral. However, the only internal test that we can make gives results completely consistent with our classification.
DISCUSSION
We have used the combination of a multiple sequence alignment of orthologous BRCA1 sequences and a measure of the chemical difference between the amino acids present at individual residues in the sequence alignment to classify missense variants and in-frame deletions detected during mutation screening of the human BRCA1 gene. From a database of sequence variants found through full sequence testing of 20 000 individuals at Myriad Genetic Laboratories, the vast majority of whom had either a personal or a family history of breast or ovarian cancer, or both, a total of 314 different missense changes and eight in-frame deletions were observed. Of these, only 21 missense changes were classified as deleterious or suspected to be deleterious and 14 as neutral or of little clinical significance before this study. In the present analysis we were able to classify an additional 50 missense variants and two in-frame deletions as probably deleterious and 92 missense variants as probably neutral. An internal test of the classification, based on the observation that it is extremely unlikely for one individual to carry two deleterious mutations in BRCA1, is completely consistent with our classification.
Both the informativeness of the multiple sequence alignment used in our analysis and the method of classification bear some discussion. Two previous studies have looked at Human missense changes in BRCA1 relative to full length multiple sequence alignments of orthologous sequences.27,30 In those studies, the authors demonstrated that known deleterious and neutral missense variants in Human BRCA1 fit the pattern of cross species sequence alignment that we have described here, but did not use the alignments to make specific predictions about unclassified variants. More recently, unclassified variants in exon 11 of BRCA1 were analysed in the context of an alignment of 57 eutherian mammal exon 11 sequences.51 From the 139 missense variants that were analysed, a set of 41 missense variants was prioritised for further study because the analysis predicted that they would affect protein function. Of these 41 missense variants, 30 appear in the Myriad Genetic Laboratories database, for a total of 118 observations. Eight of the 118 carriers of one or another of these missense variants (one each for eight different missense variants) also carried a clearly deleterious mutation in BRCA1, which is insignificantly different from the frequency of deleterious mutations in the overall test population. So it is probable that a number of these prioritised missense variants are unlikely to be deleterious, probably because the cladogram underlying that sequence alignment does not sample sufficient evolutionary time to eliminate most weakly conserved invariance.
By creating a BRCA1 alignment that incorporates two relatively diverged sequences, BRCA1s from the amphibian Xenopus laevis and the teleost fish Tetraodon nigriviridis, we have greatly decreased the number of invariant positions and presumably increased the informativeness of the alignment. However, there is neither an established criterion for informativeness of a multiple sequence alignment for applications in medical genetics nor an obvious way to know whether there is an optimum amount of sequence divergence to capture in such an alignment. The goal in creating the alignment is to minimise the number of positions in the alignment that are weakly conserved invariant rather than invariant due to shared ancestry and natural selection driven sequence conservation (invariant Class 2), while simultaneously minimising the number of positions in the alignment where conservation has been lost because second site mutations have allowed what would otherwise be an obligate conserved position to become permissive of sequence variation. Accordingly, in the best possible alignment, we would observe missense substitutions at all Class 3 positions, while all Class 2 positions would remain invariant. Of course, these two goals will not often be achieved simultaneously. In the current alignment we estimate that there are 281 Class 2 positions, out of which 125 are invariant; of 1582 Class 3 positions, only 12 are invariant. However, even though our sequence alignment is sufficiently informative that <10% of the invariant positions are from Class 3 positions, we still should expect some errors in classifying missense variants.
In fact, one variant that falls into the new group of suspected deleterious mutations, P1238L, has been seen with a known deleterious mutation (5385insC) in a BRCA1 sequence analysis not included in this study. One explanation for this observation is the possibility that P1238 is a weakly conserved invariant residue and when additional species are added to the alignment, it will no longer be an invariant. Alternatively, P1238L may be a deleterious mutation that occurs on the same allele as the known deleterious mutation.
To classify missense variants, we constructed two categories that we consider likely to identify deleterious variants and two that we consider likely to identify neutral variants. The first deleterious category, missense variants observed at positions that are invariant in the multiple sequence alignment, seems quite natural. This is because we have sampled a sufficient phylogenetic breadth of BRCA1 sequences that the number of positions in the alignment conserved due either to chance alignment or weakly conserved invariance, rather than shared ancestry and selection driven conservation, is quite small. Thus the main problems here are only that there may still be a few weakly conserved invariant positions, and that we must wonder whether very conservative missense changes at conserved positions really always confer a high risk genotype. The second deleterious category, missense variants with high chemical difference scores observed at positions in the alignment where the pairwise chemical difference scores are small, is a bit more arbitrary. Criticisms based on conservation due to chance certainly apply. In addition, our choice of a minimum ratio for the within alignment pairwise chemical difference scores to the missense variants chemical difference score of 3.0 is completely arbitrary; both the exact definition of the category and the best numerical value of the difference score cutoff could be explored further. One could also consider constructing other related categories, either stricter or more permissive, that would certainly be enriched for deleterious variants, but to a different degree.
The first category of neutral variants, missense substitutions that lead to the consensus BRCA1 sequence of another species, also seems quite natural. Here, the main problem seems to be that there are regions in the alignment, especially in sequence segments between the strongly conserved sequence elements, where the alignments of Xenopus or Tetraodon sequence are of very low quality and very gapped. Thus some of the matches between Human missense variants and Xenopus or Tetraodon sequences are probably due to chance. In addition, the likelihood should be considered that a missense change in Human sequence can actually be deleterious if it occurs at a position where other species sequences are probably deleted relative to Human. The second category of neutral variants, missense substitutions where the chemical difference score for the Human missense variant is less than one third of the highest chemical difference score of the interspecific amino acid sequence variation at that position, is also somewhat arbitrary. The criterion can be subject to the same criticisms as the first neutral variant criterion, and is also subject to the criticism that there is no concrete basis for the selected maximum ratio of within alignment pairwise chemical difference scores to the missense variant’s chemical difference score. Another limitation is that synonymous substitution matrices are not perfect, especially in their scoring of substitutions involving amino acids that have strong context specific characteristics such as cysteine or proline.
The process used here to classify missense changes proceeds from a different point of view than that which has been used for classification of sequence variants in the past. For the most part, past classification strategies have looked at sequence variants individually and arrived at a call that applies only to the individual variant. Here, we have used a series of criteria to identify groups of sequence variants and then asserted that most of the variants in a specific group are deleterious or most of the variants in a specific group are neutral. Improving the classification becomes a matter of refining the criteria for membership in a group, perhaps creating new groups, and making accurate measures of the likelihood that a member of a particular deleterious group is actually a false positive or that a member of a particular neutral group is actually a false negative. In the end, sequence variants are still classified with the disclaimer that one must always recognise the possibility of a false positive or false negative. However, one should take into account that while the combined frequency of mutations classified here as likely to be deleterious is only 0.6% among tested individuals; it constitutes almost 7% of the people found to carry a clearly deleterious mutation.
For some time, Myriad Genetic Laboratories has actually been applying a five tiered system for classifying sequence variants at splice junctions. The five tiers are deleterious, suspected deleterious, unclassified, favour polymorphism, and neutral or of little clinical significance. The suspected deleterious and favour polymorphism categories were created to provide a category for splice junction variants that on the basis of sequence analysis seem likely or unlikely, respectively, to interfere with mRNA splicing, but for which there is neither biochemical nor human genetics evidence to corroborate increased or decreased risk. We suggest that the same five tiered classification be extended to missense variants. On discovery, a novel missense variant would initially be regarded as unclassified. If analysis through the multiple sequence alignment and chemical difference score system resulted in evidence that the variant is either deleterious or neutral, then it would be classified as suspected deleterious or favour polymorphism, respectively. Either corroborating or contradictory biochemical or human genetics data might then be used to reclassify the missense variant. In some sense, the favour polymorphism category is particularly important. There are a number of ways that a missense variant in BRCA1 could be deleterious without interfering with the function of the BRCA1 protein in itself. For instance, the nucleotide substitution underlying a missense variant could interfere with a splice junction, activate a cryptic splice junction, or interfere with a member of the recently identified degenerate class of sequence elements termed exonic splicing enhancers.52 It is also possible that a missense change might create a proteolysis site or otherwise interfere with the half life of the BRCA1 protein. Any of these eventualities might result in a missense variant that looks innocuous on the basis of the analysis presented here but is actually deleterious. The category favour polymorphism provides the level of caution these possibilities require, which is not achieved by the neutral or of little clinical significance category. The long term utility of this approach will become clearer with further scientific and clinical validation.
As a stand alone method, our approach to classification of missense variants has its strengths and weaknesses; we expect that a more informative multiple sequence alignment combined with a further developed classification algorithm will move our approach closer to clinical applicability. Additionally, this work can be viewed as one component of a multimodal strategy to manage currently unclassified sequence variants better. Classification could be based on a combination of analyses, such as goodness of fit of missense substitutions within the available crystal structure of protein domains; results of available functional assays of protein domains; improved statistical analyses of splice junction fitness; robust segregation analyses when possible in the small, incompletely ascertained families undergoing clinical testing; and creatively formatted association studies designed to look at classes or groups of individually rare sequence variants. We expect that the approach we have taken will become an important component of a broad method designed to reach a definable criterion of strength of evidence that will result in clinically useful calls even though no single classification method reached the required level of confidence.
We have proceeded through this entire analysis as if all sequence variants are actually members of one of two categories, high risk or neutral. In fact, there are no data to argue against the hypothesis that some missense changes in BRCA1 confer an odds ratio intermediate between that of a high risk protein truncating mutation and a true neutral variant. If such missense substitutions, which might best be called modest or moderate risk, exist, then they should be enriched in the set of sequence variants that remain unclassified despite our best analyses. As we reach the point where we can tentatively classify ≈50% of missense substitutions observed during clinical testing of BRCA1 as either favour deleterious or favour neutral or of little clinical significance, we should prepare ourselves to consider another level of complexity in the role that this gene plays in cancer susceptibility and develop methods to meet the challenge of translating that information to clinical practice.
REFERENCES
Footnotes
-
Conflict of interest: most of the authors are employees of Myriad Genetics, Inc. This manuscript bears on their commercial test for mutations in BRCA1.