Article Text

Abstract
Introduction: It has been estimated that cytogenetically visible rearrangements are present in ~1% of newborns. These chromosomal changes can cause a wide range of deleterious developmental effects, including mental retardation (MR). It is assumed that many other cases exist where the cause is a submicroscopic deletion or duplication. To facilitate the detection of such cases, different techniques have been developed, which have differing efficiency as to the number of loci and patients that can be tested.
Methods: We implemented multiplex amplifiable probe hybridisation (MAPH) to test areas known to be rearranged in MR patients (for example, subtelomeric/pericentromeric regions and those affected in microdeletion syndromes) and to look for new regions that might be related to MR.
Results: In this study, over 30 000 screens for duplications and deletions were carried out; 162 different loci tested in each of 188 developmentally delayed patients. The analysis resulted in the detection of 19 rearrangements, of which ~65% would not have been detected by conventional cytogenetic analysis. A significant fraction (46%) of the rearrangements found were interstitial, despite the fact that only a limited number of these loci have so far been tested.
Discussion: Our results strengthen the arguments for whole genome screening within this population, as it can be assumed that many more interstitial rearrangements would be detected. The strengths of MAPH for this analysis are the simplicity, the high throughput potential, and the high resolution of analysis. This combination should help in the future identification of the specific genes that are responsible for MR.
- mental retardation
- high resolution
- high throughput
- genomic copy number changes
- BAC, bacterial artificial chromosome
- CGH, comparative genomic hybridisation
- FISH, fluorescent in situ hybridisation
- MAPH, multiplex amplifiable probe hybridisation
- MCA, multiple congenital abnormalities
- MLPA, multiplex ligation dependent probe amplification
- MR, mental retardation
- SMS, Smith-Magenis syndrome
Statistics from Altmetric.com
- BAC, bacterial artificial chromosome
- CGH, comparative genomic hybridisation
- FISH, fluorescent in situ hybridisation
- MAPH, multiplex amplifiable probe hybridisation
- MCA, multiple congenital abnormalities
- MLPA, multiplex ligation dependent probe amplification
- MR, mental retardation
- SMS, Smith-Magenis syndrome
The evolution of the human genome has resulted in a mixture of large and small interspersed and tandem segmental duplications throughout the genome. Such duplications provide substrates for homologous recombination, and consequently, the intervening regions show a considerable rate of rearrangement.1–3 Many of these rearrangements occur in regions where a change in gene dosage does not affect human health. However, after the description by Lejeune of trisomy 21 in Down’s syndrome,4 and the many subsequent publications on different aneuploidies, it became clear that the genome contains many loci for which the correct copy number is critical for normal development. Change in genetic dosage of one or more genes is one of the most common causes of mental retardation (MR). Examples of known important loci include the subtelomeric regions and the areas involved in microdeletion syndromes.
The subtelomeric regions, localised proximal to the telomeres, have been found to be especially susceptible to copy number changes, owing to repeat rich sequences that show a high frequency of recombination.1 It has been hypothesised that about 6% of the patients with idiopathic MR will have a subtelomeric rearrangement,5 a figure confirmed in several studies that have reported a frequency of 2–9% of cryptic rearrangements in MR patients.6,7
The cause for MR is only established in approximately 50% of cases, limiting the efficiency of genetic counselling, detection of carriers, and prenatal diagnosis in these families. This rather low percentage of diagnosis may have several explanations. A routine cytogenetic analysis gives a minimum resolution of only 4–10 Mb. Fluorescent in situ hybridisation (FISH) largely overcomes this limitation of resolution; however, it can only be applied to simultaneously test a limited number of chromosome regions. FISH is therefore mostly used to confirm well recognised microdeletion syndromes in patients who present a suggestive phenotype. Another potential explanation is that the genome contains undiscovered loci that are involved in the aetiology of MR. New technologies, such as multiplex amplifiable probe hybridisation (MAPH),8 multiplex ligation dependent probe amplification (MLPA),9 and array based comparative genomic hybridisation (array CGH),10 have recently been developed to search for such undiscovered regions. We chose to implement a high resolution, high throughput, rapid, and simple method, MAPH,8 which allows the simultaneous screening at the exon level for copy number changes of 40–50 different chromosomal loci in up to 96 patients in one assay.
Hollox et al11 previously described subtelomeric screening using MAPH of patients with a developmental delay. In our study, we screened loci known to be involved in MR (subtelomeric/pericentromeric regions and genes involved in microdeletion syndromes) as well as interstitial genes randomly spaced throughout the genome. A total of 30 000 gene dosage screens were performed from 188 cases with unexplained developmental delay that were each scanned for copy number changes at 162 loci. We were able to detect subtelomeric, pericentromeric, and interstitial rearrangements in a group of patients with MR and dysmorphic features and/or multiple congenital abnormalities, as well as in patients selected solely on the basis of developmental delay.
SUBJECTS AND METHODS
Probe design and MAPH
The probe design has been previously described,12 using unique sequences only. The primers of the chosen sequences were designed using Prophet (http://www.basic.nwu.edu/biotools/prophet.html), and supplied by Invitrogen Life Technologies. Products were amplified from genomic DNA by PCR and cloned into the pGEM-T easy vector (Promega). The correct insert was confirmed by sequencing with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems) at the Leiden Genome Technology Center, using an ABI 3700 Sequencer (Applied Biosystems).
MAPH was performed as described by White et al12 (see also Leiden Muscular Dystrophy Pages (http://www.dmd.nl/DMD_MAPH.html)).
Study population
The DNA of 188 patients (110 males and 78 females) from the Center for Human and Clinical Genetics Leiden (a DNA diagnostic laboratory) was analysed. The patients had been seen by a clinical geneticist or a paediatrician and diagnosed with developmental delay. The study population was divided into two groups. The first group contained 123 coded patients who had been referred for fragile X screening. Before testing, information about the results of additional tests, such as karyotyping, was not known to the investigators. The second study group (n = 65) was known to have a normal karyotype and had tested negative for fragile X screening. All patients had (multiple) congenital malformations or dysmorphic features in addition to psychological developmental delay.
Data analysis
The data were analysed with GeneScan Analysis and Genotyper Software (Applied Biosystems). These programs provide information about the length, peak height, and peak area of the DNA fragments. Peaks were not used for analysis if they were outside predefined thresholds (upper and lower limits of 12 000 and 150 units, respectively). To obtain a ratio, the height of a given peak was divided by the sum of the heights of the four nearest peaks. As it is not likely that all four probes from diverse regions of the genome are altered in one patient, adding unrelated standards was not necessary in most of the probe sets. For the chromosome 22 probe set, however, unrelated probes, containing sequences from other chromosomes, were used as references.
The median ratio for each probe within a single hybridisation (minimum number of samples 8; maximum number 12) was determined and used to calculate a normalised ratio for each patient. Within each patient, initial “normal” thresholds were set as 0.75 and 1.25. The standard deviation from the ratios within these limits was calculated, and three times this standard deviation was used as the threshold for any given patient. Any probe that was outside these limits was retested, and samples that showed an apparent copy number change in duplicate were examined further using other techniques. Samples that showed a standard deviation of >10% over probes within the normal thresholds were retested.
Verifying the MAPH results
Copy number changes detected by MAPH were verified using another technique, primarily FISH with a bacterial artificial chromosome (BAC) or cosmid probe covering the appropriate genomic region. The BACs used were designed by Flint,13 or supplied by Vysis Abbott Laboratories (TV, Telvysion, LSI, locus specific identifiers) or selected from the RPCI human BAC library. The FISH experiments were performed following standard operating procedures as described in Dauwerse et al.14 Some MAPH results were verified using MLPA.9
RESULTS
Genotyping
We designed several probe sets covering both the subtelomeric/pericentromeric and interstitial regions, including genes involved in microdeletion syndromes, genes on chromosome 22, and genes spread across all chromosomes (table A, supplemental). The subtelomeric probe set is composed of probes corresponding to the 41 subtelomeric regions, preferably an exon of a gene within 1 Mb from the telomere, five genes near the centromere on the q arm of the acrocentric chromosomes, a sequence in the pseudoautosomal region of chromosome Xq and Yq, and an exon of a Yp specific gene. The microdeletion probe set was made up of 27 probes from 21 different genes involved in microdeletion syndromes (Williams, Prader Willi, Angelman, Smith-Magenis, Sotos, 22q11, Alagille, and Wolf-Hirschhorn syndromes). The chromosome 22 probe set included 19 probes from genes on chromosome 22 with approximately 1 Mb spacing. Finally, we used two probe sets containing a total of 68 interstitial genes spread throughout the genome.
We applied these probe sets following two methods of validation. Firstly, a probe was considered to be reliable when the standard deviation over 12 unaffected samples (one hybridisation) was <15%. Secondly, where possible, we verified the unique and correct localisation of the probes using DNA from patients with known aberrations (42% of the subtelomeric probes, 70% of the microdeletion probes).
Overall, 188 patients were screened for deletions and duplications at 162 loci, resulting in the detection of 19 copy number changes. Of these, four aberrations turned out to be cytogenetically visible, namely an isochromosome 18p (karyotype 47, XY, +i(18p)), a marker chromosome (karyotype 47, XY, +mar.ish der(22)t(8;22)(q24.1;q11.2)), a triple X female (karyotype 47, XXX) and a Turner syndrome (karyotype 45, X), because the outcome of additional investigations had not been made known to the investigators before testing. These patients and their corresponding aberrations were not included in the calculation of the percentage of rearrangements found by MAPH; however, they emphasise the usefulness of MAPH for detecting copy number changes.
In total, eight subtelomeric/pericentromeric rearrangements were found (table 1; upper part). Five of these mutations were detected in the group of MR patients with additional dysmorphic features or additional congenital malformations (5/65 = 7.7%) and the remaining three subtelomeric aneusomies were diagnosed in the group selected on the basis of developmental delay only (3/123 = 2.4%). The smallest mutation found was a deletion of 110 kb maximum present in chromosome band 7p22.3 (table 1, F; and data not shown). Seven rearrangements were interstitial mutations. These are summarised in the lower part of table 1. Where possible, the DNA of both parents of these patients was tested; 75% (9/12) were shown to be de novo. The duplication of 14q11.2 (table 1, O) and the 7ptel deletion (table 1, F) were also found in the parental DNA, and one of the parents of patient E was a balanced translocation carrier.
An overview of all 15 patients (A–O) with MAPH detected subtelomeric/pericentromeric and interstitial aneusomies. After the verification of these imbalances by FISH or MLPA, the karyograms of the patients were re-examined at a resolution of 500–550 bands. The results obtained are shown in the column ‘cytogenically visible’. The clinical features known to be related to the rearrangement found by MAPH are highlighted. The presence or absence of a genotype–phenotype correlation is summarised under “Pathogenic”.
As the number of cytogenetically detectable aberrations is highly dependent on the banding resolution, the karyograms of all 15 patients with a MAPH detected rearrangement were re-examined. At a resolution of 500–550 bands per haploid set, the karyograms showed that two subtelomeric copy number changes should have been detected cytogenetically (table 1; A, C). The detection of a 1ptel deletion (table 1, H) was doubtful; however, the duplication of 1ptel (table 1, H) was picked up. This implies that although the presence of the copy number change was known, 63% (12/19) of these genomic changes found in this study were cytogenetically undetectable using karyotyping at a resolution of 500–550 bands.
Case descriptions
Case 1
This 15 year old girl was diagnosed with total anomalous pulmonary venous return, hearing loss in combination with a narrow external auditory meatus, and MR. Physical examination at the age of 14 years showed a short stature (−3 SD) and some facial dysmorphic features (small palpebrae, broad mouth, thin upper lip). Karyotyping at a resolution of 400 bands and FISH studies of the 22q11 region did not detect any rearrangements. MAPH study showed a de novo deletion of the subtelomeric region of 18q, which was confirmed by FISH using probe TV18q. The clinical features of this patient are consistent with those of the 18q syndrome phenotype.15
Case 2
A male patient, who had previously tested negative for Williams syndrome, was diagnosed with a de novo deletion of 16ptel by MAPH. FISH analysis confirmed this finding and limited the proximal breakpoint to chromosome band 16p13.3, distal to the PKD1–TSC2 (LocusLink 5310–7249) gene cluster16 using probe COS15A. As expected, owing to the location of the alphaglobin gene (HBA1; LocusLink 3039) in this region (16p13.3),17 further investigation showed that this patient had mild anaemia (alpha thalassaemia heterozygosity) in addition to his moderate mental handicap and dysmorphic features.
Case 3
This boy was seen by a clinical specialist at the age of 2.5 years for his psychomotor retardation and joint hyperflexibility. Physical examination showed few dysmorphic features (a tent shaped mouth), hypotonia, and hypermobility. MAPH analysis revealed a de novo deletion within chromosome band 17p11.2 corresponding to the Smith-Magenis syndrome (SMS) region, using a probe for the DRG2 gene (LocusLink: 1819). The more distally located COPS3 gene (LocusLink: 8533) showed two copies (fig 1a). Additional MLPA testing showed that the RAI1 gene (LocusLink: 10743) was also deleted in this patient (fig 1b), and FISH analysis (probe LSI-SMS) verified the deletion of part of chromosome band 17p11.2 (fig 1c). Recently, three dominant frameshift mutations in RAI1 have been identified in three patients with phenotypic characteristics of SMS but no cytogenetically detectable deletion of chromosome band 17p11.2.18 The authors argue that mutations in RAI1 are responsible for most of the characteristic features of SMS and that further variation is caused by hemizygosity of the other genes in the chromosome region.

The plots correspond to the MAPH results showing (A) a deletion of the DRG2 gene, two normal copies of COPS3A (RAI1 not present), and the MLPA results; and (B) a deletion of RAI1, a deletion of DRG2, and a normal ratio of COPS3A. (C) The additional FISH analysis using the LSI-SMS probe specific for the Smith Magenis chromosomal region shows a normal signal on the short arm of only one copy of chromosome 17.
Case 4
This male patient showed at the age of 12 years a mild learning disability, a low voice, a disproportionally short stature (height −2 SD, span −3 SD for height, sitting height −0.5 SD, head circumference −2 SD), limited elbow extension, a permanently extended, inflexible fifth digit of both hands with a ram’s horn shaped nail and hypotrophy of the hypothenar muscles (fig 2), and a short broad great toe on both feet. The hand x ray revealed short metacarpals I and V, short distal phalange V, and a delay of bone maturation. In this patient, a de novo deletion of 4q34.1 was detected and confirmed by FISH (probe RP11-475B2). Analysis with a more distally located MAPH probe at chromosome band 4q35.1 showed that this latter region was still present, indicating an interstitial rearrangement. Additional FISH experiments using different BAC probes limited the deletion to a maximum of 3 Mb (data not shown).

The right hand of case 4 showing a short, inflexible fifth digit with a ram’s horn shaped nail and hypotrophy of the hypothenar muscles.
Patients with an interstitial 4q deletion have been described with a range of features, depending on the proximal and distal breakpoints of the deletion.19 As it is known that fifth finger anomalies and short stature are found in patients with an interstitial deletion of 4q including 4q34,20 as well as in patients with a terminal deletion of 4q, it is possible that the genes responsible for these features are located within this region.
Case 5
This mildly retarded man, with a de novo duplication within chromosome band 20p12.2, containing the Jagged1 gene (JAG1; LocusLin: 182), died at the age of 60 years from multiple myeloma. He had been institutionalised for over 40 years in a psychiatric hospital because of aggressive behaviour, and was diagnosed as schizophrenic. To the best of our knowledge there has been only one previous report21 of a duplication of 20p11.21–p11.23, in four members of a family with clinical signs of Alagille syndrome. As our patient is not available for further investigation, it remains unclear whether he had such features.
Case 6
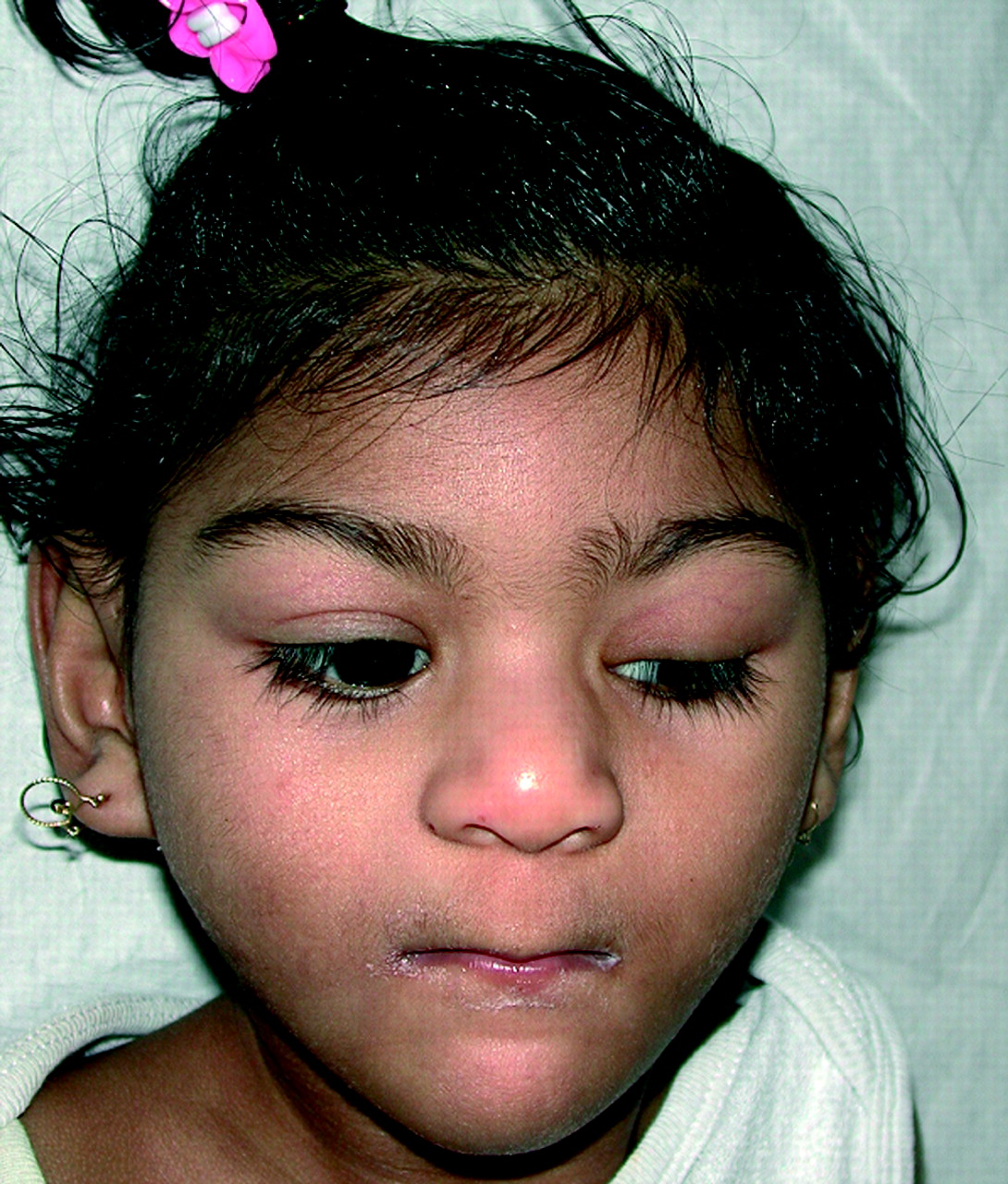
After 41 weeks of gestation, this child was born with a birth weight of 1995 g (⩽2.5 SD) and a head circumference of 28.5 cm (⩽2.5 SD). At the age of 25 months, her psychomotor development was severely delayed and she suffered from epilepsy. Physical examination showed growth retardation (length ⩽2 SD; weight −6 SD), microcephaly (head circumference −6 SD), hypertonicity, dystonic movements, facial dysmorphisms (ptosis of the left eye, flat philtrum, thin upper lip; fig 3) ear pits, café au lait spots, and absence of the labia minora. Further investigation revealed corpus callosum hypoplasia and deformed gyri, the presence of only one kidney and mildly increased urinary glutaric acid.

{kind=link}
{kind=link}
{kind=link}
Facial dysmorphism of case 6. Note the microcephaly, ptosis of the left eye, flat philtrum, and thin upper lip.
Using the microdeletion probe set, a duplication of 22q11.2 was detected by MAPH, and FISH analysis in interphase nuclei confirmed this finding (LSI TUPLE1). The patient’s mother did not carry the duplication, and the father was unavailable for testing. We plan to use polymorphic markers to determine the parental origin of the aberrant chromosome 22.
DISCUSSION
Using MAPH analysis, we performed a high resolution duplication/deletion screening of 188 patients with a developmental delay; 162 loci per patient were tested, amounting to over 30 000 typings. The MAPH probes designed for this study can be broadly divided into two groups: (a) subtelomeric and pericentromeric probes (n = 48) and (b) interstitial probes (n = 114), containing sequences located in regions previously found to be rearranged in mentally retarded individuals, and genes randomly spaced through out the genome.
We detected 4.3% (8/184) subtelomeric/pericentromeric rearrangements (six deletions, one duplication, and one subtelomeric deletion/duplication in one patient), using 48 MAPH probes. A subdivision of subtelomeric aberrations over our two study populations agrees with the findings of Knight et al22 and Yasseen et al.23 The percentage of subtelomeric mutations detected was higher in a group of MR patients with additional malformations (7.7%) than in a group selected on the basis of developmental delay only (2.5%). This supports the suggestion of De Vries et al that pre-selection of patients for subtelomeric screening is worthwhile. However, pre-selection of these patients for subtelomeric rearrangements is difficult, as only two clinical features (perinatal onset growth retardation and a positive family history) differed significantly between patients with subtelomeric aneusomies and patients with idiopathic MR.24 Our overall percentage is similar to that reported in a recent paper that summarised all previous subtelomeric publications.7 A total of 131 subtelomeric imbalances were found using several different methods among 2582 MR patients, resulting in an overall frequency of 5.1%. A review of the corresponding clinical aspects of these subtelomeric rearrangements has been published recently.25 After re-examining the karyogram of our patients at a banding resolution of 500–550 bands, it showed that five MAPH detected subtelomeric imbalances were not cytogenetically visible, despite the knowledge of a copy number change present. This means that the percentage of “true” submicroscopic subtelomeric/pericentromeric findings is ~3% (5/184) in this study.
Previous reports by Sismani et al26 and Hollox et al11 had already shown the ability of MAPH to detect subtelomeric copy number changes. Hollox et al found a copy number change in 5 of 37 male patients (13.5%) who had been referred for fragile X screening. The higher percentage of mutations found by this group may be due to differences in selection criteria for fragile X screening.
We also screened the subtelomeric/pericentromeric regions in eight newborns suffering multiple congenital abnormalities (MCA). Among these patients, one deletion of the subtelomeric region of chromosome 15 was detected and subsequently confirmed by FISH (data not shown).27 To determine whether it is worthwhile to test this group for submicroscopic mutations, more newborns with MCA should be examined. The ease and relatively low cost of the MAPH technique means that such analysis is feasible. Moreover, new techniques such as MAPH/MLPA and array CGH provide the possibility of genetic diagnosis at a younger age. As the suggestive phenotype for some microdeletion syndromes emerge only later in life, this diagnosis would be very important for providing appropriate healthcare.
In addition to the reports published by Sismani et al25 and Hollox et al,11 we also examined interstitially localised genes, including genes involved in several microdeletion syndromes, genes on chromosome 22 (as this was the first chromosome to be completely sequenced), and genes that are spread throughout the genome and might be involved in cognitive development. Recently, Bailey et al3 argued that regions between highly similar duplications (low copy repeats) are prone to recombination and consequently, copy number changes occur at a higher frequency in these regions compared with other loci in the genome. Several of the areas described were also tested in this study, mostly corresponding to chromosomal regions involved in microdeletion syndromes. In total, seven interstitial deletions and duplications were detected, of which five were diagnosed in three different regions known to be involved in the microdeletion syndromes and flanked by segmental duplications. Three of these interstitial rearrangements detected include duplications of regions that are usually deleted (the chromosome regions of Smith Magenis (17p11.2), DiGeorge (22q11.2), and Alagille syndromes (20p12.2)). This observation supports the theory that the regions between low copy repeats can both be deleted and duplicated, and implies that the number of patients suffering from a microduplication syndrome is currently probably underestimated. The phenotype (if any) of a microduplication syndrome might, however, be less severe, and under standard diagnostic conditions, the detection of duplications is more problematical. It should be noted that in the second study group, the cases with a distinctive phenotype for a specific microdeletion syndrome were not included.
As has been the case during the development of every new technique, the genomic variations detected can be divided into the following subclasses: (a) genetic changes that are clearly pathogenic, (b) rearrangements that may or may not be causal to the patient’s problem, and (c) polymorphic changes. In some cases, extensive clinical studies will be needed to determine to which category a newly detected aberration belongs. In two of our cases, we could detect the rearrangement in one of the parents (the duplication of chromosome band 14q11.2 and the deletion of 7ptel on chromosome band 7p22.3). One explanation is that these imbalances are polymorphic, and that the phenotype of the patient is not related to the copy number change. However, other explanations are possible: (a) the affected region is imprinted, and the parental origin of this region is critical in causing the deleterious phenotype;28 (b) allelic variation in the expression of the genes may influence the phenotype;29 and (c) low penetrance of the rearrangement—that is, a genetic defect does not always lead to a phenotypic effect. The detection of such rearrangements will increase as high resolution techniques are applied, and this will pose new problems for genetic counselling. Therefore, it is important to map these familial imbalances in further detail to allow a genotype–phenotype correlation in larger populations of individuals with the same copy number change. In this way, the understanding of any clinical consequence of such a rearrangement should be improved.
Based on previous publications, seven rearrangements found in this study were considered to be pathogenic (table 1). In the remaining cases, the data available in literature were insufficient to support a conclusion that the aneusomy detected is related to the phenotype of the patient. It should be noted that the fact that a rearrangement is de novo is not in itself proof that it is causally related to the deleterious phenotype.
Several different methologies have been described to identify changes using MAPH and MLPA. These include visual comparison of traces from controls and patients,30 the setting of arbitrary thresholds,26 and bivariate analysis.11 We observed that the standard deviations for each probe varied slightly between hybridisations, and could be normalised only within a single hybridisation. The standard deviation of “normal” probes within each patient was calculated, with 3 times this figure defining the threshold for a potential rearrangement, thus minimising the effect of any genuine copy number changes on the analysis. As false negative results are, by definition, mutations that were not detected, it is difficult to determine the percentage. To gain an estimate as to the actual false negative rate, we looked at a number of samples where a mutation was previously known. We tested 30 samples that had aberrations at loci corresponding to 39 of the probes used. The appropriate copy number changes were detected in all cases. Using the LaPlace formula p = (x+1)/(n+2) to provide a false negative rate from our data yields an expected value of ~2.5%. This figure suggests that the true false negative rate would be, at least for the 39 probes examined, comparable to the 2% theoretically predicted by Hollox et al.11 Of course, it would be desirable to test all the probes on known mutations in the future.
The number of interstitial aneusomies found in this report strengthens the arguments for genomewide screening for copy number changes in developmentally delayed patients. In most clinical laboratories, deletions and duplications are detected by FISH. This usually focuses on only one region per hybridisation, and is therefore relatively slow and expensive. Several new technologies have emerged that facilitate large scale and genomewide screening of deletion and duplication mutations. For genomewide screening, array CGH currently seems to be the most attractive, with recent publications describing screening with approximately 2000 BAC-PAC clones at an average resolution of 1.5 Mb.31,32 This is impressive, but inherently means that 90% of the genome is not screened. In addition, probes in array CGH are 100–200 kb BAC clones, often covering more than one gene and thus able to pick up large multi-gene deletions/duplications only—that is, those >100 kb, while it is probable that a significant proportion of deletion/duplication mutations are smaller than this. In contrast, it is possible to detect rearrangements of only 100 bp using MAPH and MLPA technology. By applying a high resolution method, however, the percentage of the genome that can be screened using the same number of probes will be much less compared with array CGH. Using MAPH/MLPA, it is not possible to screen the whole genome for copy number changes at this moment, unless a very large number of probes are included. For this reason, a different approach is required. We consider array CGH to be an excellent tool for finding large regions in the genome where genes involved in particular diseases reside. As soon as these areas have been identified, targeted and much cheaper assays can be designed, zooming in on these regions only. For these reasons, we believe that gene specific screening is ultimately more attractive. With that in mind, MAPH/MLPA have an important role in such analyses, as they are able to pick up both large and small deletions/duplications.
Acknowledgments
We would like to thank all physicians of the Center for Human and Clinical Genetics Leiden for selecting patients and gathering blood samples, Dr J P Schouten (MRC-Holland, Amsterdam) for providing the MLPA probes, the Leiden Genome Technology Center for technical assistance, Dr P Eilers and E Sterrenburg for giving statistical advice, Dr C Rosenberg for critical reading of the manuscript, Dr E Peeters for her efforts, and the patients and families for their cooperation. M Kriek is funded by ZON-MW (AGIKO fellowship 940–37–032).
REFERENCES
Footnotes
-
↵* Department of Clinical Genetics, University Hospital Groningen, The Netherlands
-
The first two authors contributed equally to this work