Article Text

Abstract
Background: Germline mutations in mismatch repair genes, mainly in hMLH1, hMSH2, and hMSH6, predispose to the hereditary non-polyposis colorectal cancer (HNPCC) syndrome. A substantial fraction of these mutations exists in genomic rearrangements of hMSH2 and hMLH1. In contrast, genomic rearrangements have not been reported in hMSH6.
Methods: Out of 15 HNPCC or HNPCC-like patients who developed tumours with loss of hMSH6 protein expression, we selected three patients who still had no germline mutations after gene sequencing. Genomic DNA of these patients was analysed using PCR based relative quantification of hMSH6 fragments. Indicated exon deletions and amplifications were characterised by long range PCR and sequencing.
Results: Genomic rearrangements were identified in two of the three patients. Breakpoint analyses showed an Alu repeat mediated deletion of 13.0 kb affecting the promoter region, exon 1, and exon 2 in one patient, and a duplication of 4.9 kb containing 1.6 kb of the 3′ end of exon 4 and exon 5, integrated into intron 5, in the other patient.
Conclusions: Although genomic rearrangements of hMSH6 only play a small role in the spectrum of all mutations predisposing to HNPCC, our results suggest that up to 10–20% of patients with hMSH6 negative tumours harbour germline rearrangements in this gene.
- genomic rearrangements
- hMSH6
- HNPCC
- microsatellite instability
Statistics from Altmetric.com
Hereditary non-polyposis colorectal cancer (HNPCC, MIM 114500) is a highly penetrant, autosomal dominant cancer susceptibility syndrome. Besides colorectal cancer, affected subjects are at increased risk to develop endometrial, extracolonic gastrointestinal, ovarian, and ureteral carcinoma, and brain cancer.1 A hallmark of most of these malignancies is the contraction/expansion of simple sequence motifs,2,3 termed microsatellite instability (MSI). Germline mutations in human mismatch repair (MMR) genes, almost exclusively in hMSH2, hMLH1, and hMSH6, have been found in HNPCC, or HNPCC-like cases.4–6 The average age at onset of the disease has been shown to be slightly higher in hMSH6 mutation carriers compared to hMSH2 and hMLH1 mutation carriers, which might reflect a lower penetrance of hMSH6 mutations.7,8 Preference of instability at mononucleotide repeats in hMSH6 deficient tumours has been reported by some authors,8–11 but has not been found by others.7,12
Exon by exon screening or direct sequencing of the aforementioned genes are commonly used for mutation analysis. These methods show mutations in up to 70% of HNPCC or HNPCC-like patients with tumours showing high level microsatellite instability (MSI-H). In the majority of patients with MSI-H tumours but without detectable MMR mutations, there is still strong evidence for genetic predisposition within the same MMR genes owing to the loss of protein expression in these tumours.11 Genomic rearrangements affecting hMSH2 and hMLH1, mostly deletions several kb in size, have been reported among these patients.12–17 Large deletions may comprise 30% of all hMSH2 mutations.14 Rearrangements of hMLH1 have been less frequently reported with the exception of the Finnish population, which harbours a 3.5 kb founder deletion affecting exon 16.13 No such rearrangements have been found in hMSH6.16
We have analysed the hMSH6 gene for genomic rearrangements and identified a deletion and a duplication.
MATERIALS AND METHODS
Clinical samples
From a clinical study in 231 patients with suspected HNPCC syndrome, we identified three patients with loss of hMSH6 expression along with normal nuclear hMSH2 expression in their tumours, who had no germline mutations after the sequencing of the coding region of hMSH6. These patients fulfilled the Bethesda guidelines for HNPCC18 and developed tumours classified as MSI-H owing to instability at two mononucleotide repeats (BAT25 and BAT26) and at one or none of three dinucleotide repeats (D5S346, D2S123, D17S250). Microsatellite analysis, immunohistochemistry, and sequencing were carried out as described previously.19,20
Genomic DNA from peripheral blood of these patients and four healthy blood donors were isolated, applying the QIAamp™ blood and tissue kit (QIAGEN, Hilden, Germany), and used for the analyses. Written informed consent was obtained from the patients investigated.
Relative quantification of gene fragments
Three panels of gene fragments spanning most of the coding region of the hMSH6 gene were PCR amplified. Panel 1 comprised exons 2, 3, 5, and 8, panel 2 comprised exons 1, 3, 4–5′ end, and 4–3′ end, and panel 3 included exons 2, 4-centre, 6, and 8. Exons 7 and 9–10 were not analysed separately because of their close proximity to exons 6 and 8, respectively, based on small exon sizes, small, almost Alu repeat free introns, and intronic polymorphisms in all three patients, as shown by gene sequencing (data not shown). Exon 13 of hMLH1 was included as a control. Primers are available on request.
Multiplex PCRs contained 30–50 ng DNA, 200 mmol/l of each dinucleotide, 1.7 mmol/l MgCl2, 200 nmol/l of each primer, and 1 unit of Expand High Fidelity PCR system (Roche Diagnostics, Mannheim, Germany) in a total volume of 25 μl. Conditions were 30 seconds at 94°C, 30 seconds at 58°C, and 30 seconds at 72°C for 22 to 24 cycles with five minutes at 94°C before and four minutes at 72°C after cycling. One primer for each fragment was Cy5-labelled, facilitating detection of amplified fragments on the sequencing devices. Of each PCR, 0.5–2.0 μl were electrophoresed on Automated Laser Fluorescence (ALF) express sequencing devices according to standard protocols. Fragment signals were analysed for relative quantity and height applying the ALLELELINKS™ program (both Amersham Biotech, Freiburg, Germany). Genomic DNA of healthy blood donors served as controls.
Breakpoint analysis
Long range PCRs using the Expand Long PCR system (Roche Diagnostics) were applied for amplification of rearrangement specific products. PCR products were sequenced by primer walking applying ALF express sequencing devices. All nucleotide numbering, fragment sizes, and information on repeat regions were given according to GenBank accession AC006509 containing the complete genomic sequence of the hMSH6 gene.
RESULTS AND DISCUSSION
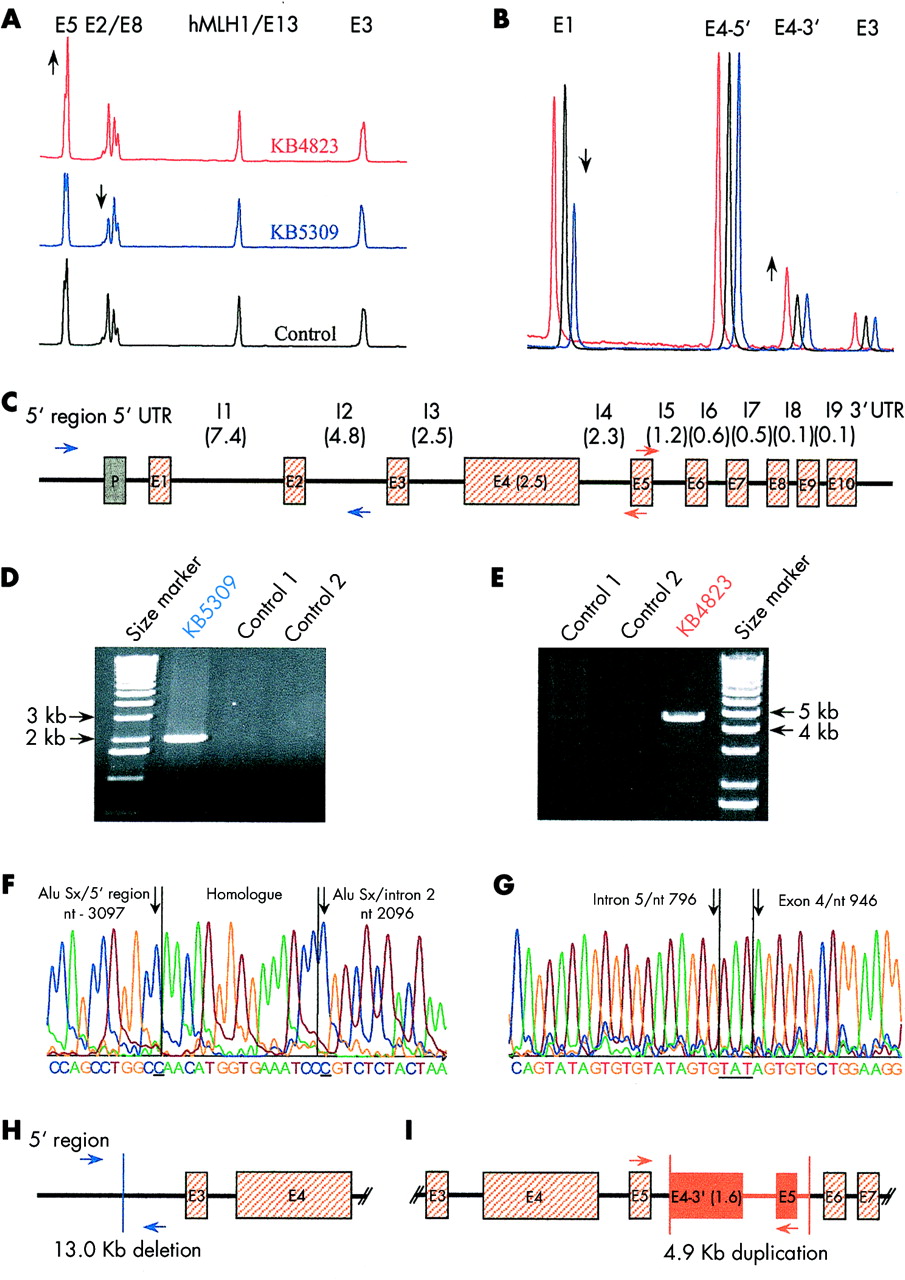
Differences in the quantity of amplified hMSH6 fragments were found in two patients after quantification of genomic fragments of the hMSH6 coding region using multiplex PCR. A reduction of signals for exon 1 and exon 2 of around 50% was found in patient KB5309 (fig 1A, B). DNA from patient KB4823 showed an increased amplification of 30–50% in the centre and the 3′ region fragments of exon 4 and of exon 5, whereas the 5′ end of exon 4 appeared to be normal, suggesting a duplication with an intra-exonic breakpoint. Measured quantities of all other fragments from patients KB4823 and KB5309, as well as all fragments amplified from DNA of the remaining patient did not indicate deletions or amplifications.
{kind=link}
Relative quantification of exon fragments of hMSH6 with (A) panel 1 and (B) panel 2, as described in the Materials and methods section. Patient KB4823 (red lines) shows increased signals for exon 5 and the 3′ end of exon 4. Patient KB5309 (blue lines) shows decreased signals for exons 1 and 2. (C) Genomic structure of hMSH6. Number in parentheses: sizes in kilobases. I: intron; P: promoter, E: exon; UTR: untranslated region. Blue and red arrows mark locations of primers used for breakpoint analyses in patients KB5309 and KB4823, respectively. (D) Long range PCR for patient KB5309, applying primers indicated in (C) (3.6 kb upstream of exon 1 and 2.5 kb downstream of exon 2), separated on an agarose gel. (E) Long range PCR for patient KB4823 applying primers indicated in (C) (primers in exon 5, where the antisense primer is located 93 bp upstream of the sense primer), separated on an agarose gel. (F) Breakpoint sequence of the deletion in patient KB5309. The underlined nucleotides are the last nucleotide specific to the Alu Sx repeat in the 5′ region and the first nucleotide specific to the Alu Sx repeat in intron 2. (G) Sequence of the integration site of the duplicated fragment of patient KB4823. The underlined nucleotides do not belong to the integration site in intron 5 or the 5′ end of the duplicated sequence (exon 4). Schematic presentation of (H) the deletion in patient KB5309, and (I) the duplication in patient KB4823, compared to (C), respectively.
Breakpoint analyses were performed in order to verify the results obtained from relative quantification of gene fragments. Long range PCRs with the sense primers located at 3.6 kb and 5.4 kb upstream of exon 1, in combination with antisense primers located at 2.5 kb downstream of exon 2 and in exon 3, only yielded amplicons with DNA from patient KB5309. Combination of the sense primer 3.6 kb upstream of exon 1 with the antisense primer 2.5 kb downstream of exon 2 (shown schematically in fig 1C with blue arrows) amplified a 2 kb fragment (fig 1D), indicative of a genomic deletion of about 13 kb affecting exons 1 and 2 (fig 1H). Sequencing of PCR products showed a deletion of 12 996 bp mediated most probably by recombination between two Alu repeats of the Sx family (fig 1F). The breakpoints were located within a 15 bp sequence that is identical in both involved AluSx repeats and comprised bases 3097 to 3082 upstream from exon 1 and bases 2010 to 2025 bp downstream from exon 2, respectively. In the DNA from patient KB4823, PCR, applying a sense primer located at exon 5 in combination with an antisense primer located a short way upstream of the sense primer in exon 5 (shown schematically in fig 1C by red arrows), amplified a fragment of approximately 4.8 kb (fig 1E). Combination of the same sense primer, together with another antisense primer located in the 3′ end of exon 4, yielded a 2.6 kb fragment. These results are suggestive of a duplication of the 3′ end of exon 4, intron 4, and exon 5, integrated into intron 5 in the sense direction (fig 1I). Sequencing of the fragments showed the 5′ end of the duplicated fragment, which contained 1600 bp of the 3′ end of exon 4 integrated at nucleotide 797 of intron 5 (fig 1G). There were three inserted nucleotides (TAT) at the 5′ end of the duplicated sequence that did not originate from the integration site in intron 5 or the 5′ end breakpoint of the duplicated sequence in exon 4. Amplification of intron 5 using primers in exon 5 and intron 6 yielded the expected 1.5 kb fragments in all patients and controls. Sequencing of 400 bp surrounding nucleotide 797 of intron 5 showed no differences from the wild type with the exception of a C/T polymorphism at nucleotide 548 of intron 5 in the heterozygous state, confirming that the fragment was amplified from both chromosomes in patient KB4823. Furthermore, PCR using a sense primer specific for the intron 5-exon 4 junction and an antisense primer in intron 8 amplified an anticipated 7.2 kb fragment in patient KB4823 only. Specificity of the PCR was verified by DNA sequencing. These results predicted a 3′ end breakpoint of the duplicated fragment identical to the integration site and, therefore, a duplication of 4926 bp. The duplication did not involve known repetitive elements, but the three nucleotides added to the 5′ end of the duplication resulted in a three times repeated ATA(GT)2/3 sequence motif at the intron 5-exon 4 junction.
Patient KB4823 suffered from synchronous carcinomas of the endometrium and the ovary at the age of 51 and had a family history of colorectal and endometrial carcinomas among first and second degree relatives. Patient KB5309 suffered from two synchronous colorectal carcinomas at the age of 54. The family history was unknown.
The deletion in the germline of patient KB5309 removed the functional promoter region 21 and the first two coding exons from one allele of the hMSH6 gene and, therefore, can be postulated as a disease causing mutation. The functional relevance of the duplication of the 3′ end of exon 4 and of exon 5 in the germline of patient KB4823 is debatable. Exon 5 is integrated completely with its splice sites and the integration of a second copy of exon 5 in the hMSH6 transcript would result in a truncated protein. No material for transcript analysis was available from this patient, but the loss of hMSH6 expression in the tumour of patient KB4823 may emphasise the pathogenic nature of the duplication.
To date, we have analysed 15 patients with hMSH6 negative tumours and have identified small insertions, deletions, and base substitutions considered pathogenic in the germline of 12 patients.11,20 Identification of genomic rearrangements in two of the remaining three patients increased the mutation detection rate to 93%. Although the numbers are low, our findings suggest that genomic rearrangements may comprise up to 10–20% of all mutations of this gene, even though it has a relatively small genomic size of approximately 20 kb.22 The frequency of hMSH6 mutations is estimated to account for around 10% of all MMR mutations.9,11 Therefore, genomic rearrangements in hMSH6 play only a small role in HNPCC predisposing germline mutations, but may play a substantial role in the hMSH6 based predisposition. According to Charbonnier et al,16 the molecular diagnosis in HNPCC should begin with the relative quantification of gene fragments of hMSH2 because of its simplicity and rapidity and the frequency by which genomic rearrangements are found in hMSH2. Based on our findings, this strategy could be extended to hMSH6, but only to patients with hMSH6 negative tumours, given the relative rarity of hMSH6 mutations.
Acknowledgments
We thank Ms A Rudek for excellent technical assistance. This work was supported by the Deutsche Krebshilfe grant “Familiärer Darmkrebs” (70-2367-Scha2).