Article Text

Abstract
Although possession of the ε4 allele of the apolipoprotein E gene appears to be an important biological marker for Alzheimer’s disease (AD) susceptibility, strong evidence indicates that at least one additional risk gene exists on chromosome 12.
Here, we describe an association of the 3′-UTR +1073 C/T polymorphism of the OLR1 (oxidised LDL receptor 1) on chromosome 12 with AD in French sporadic (589 cases and 663 controls) and American familial (230 affected sibs and 143 unaffected sibs) populations. The age and sex adjusted odds ratio between the CC+CT genotypes versus the TT genotypes was 1.56 (p=0.001) in the French sample and 1.92 (p=0.02) in the American sample. Furthermore, we have discovered a new T/A polymorphism two bases upstream of the +1073 C/T polymorphism. This +1071 T/A polymorphism was not associated with the disease, although it may weakly modulate the impact of the +1073 C/T polymorphism.
Using 3′-UTR sequence probes, we have observed specific DNA protein binding with nuclear proteins from lymphocyte, astrocytoma, and neuroblastoma cell lines, but not from the microglia cell line. This binding was modified by both the +1071 T/A and +1073 C/T polymorphisms. In addition, a trend was observed between the presence or absence of the +1073 C allele and the level of astrocytic activation in the brain of AD cases. However, Aβ40, Aβ42, Aβ total, and Tau loads or the level of microglial cell activation were not modulated by the 3′-UTR OLR1 polymorphisms. Finally, we assessed the impact of these polymorphisms on the level of OLR1 expression in lymphocytes from AD cases compared with controls. The OLR1 expression was significantly lower in AD cases bearing the CC and CT genotypes compared with controls with the same genotypes. In conclusion, our data suggest that genetic variation in the OLR1 gene may modify the risk of AD.
- Alzheimer’s disease
- polymorphism
- OLR1
- brain
Statistics from Altmetric.com
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that occurs predominantly in later life. The main pathological features of AD are characterised by neurofibrillary tangles and senile plaques caused by the progressive deposition of Aβ peptides in the brain, composed mainly of 39-43 peptides generated by proteolytic cleavage of the Aβ precursor protein (APP).
Genetic variations in four genes have been directly linked to the pathogenesis of AD. Mutations in three genes coding for the amyloid precursor protein (APP), presenilin 1 (PS-1), and presenilin 2 (PS-2) account for most cases of early onset, autosomal dominant familial AD (FAD), but only for 2% of all the AD cases.1,2 The genetics of the late onset form of the disease is far more complex and only the ε4 allele of the apolipoprotein E (APOE) gene has so far been identified as playing a major role in the causation of sporadic cases.3
Although all these genes are known or suspected to be involved in the “amyloid cascade” either by increasing Aβ production or deposition or by enhancing Aβ toxicity,4APOE differs in several aspects from these other genes and, until now, is the only gene known to be involved in the lipid pathway.5 A growing body of evidence suggests that lipid metabolism and high cholesterol levels might be linked to AD.6 Epidemiological studies indicate that people treated with inhibitors of cholesterol biosynthesis, such as statins, may have a reduced risk of developing AD.7 Interestingly, numerous in vitro and in vivo studies have shown that cholesterol modulates APP processing and affects APP mRNA expression, whereas Simvastatin has been reported to reduce strongly the level of both Aβ40 and Aβ42 peptides.6,8,9 Finally, the APOE ε4 allele has been correlated with high cholesterol level.10 ApoE is a lipid and cholesterol transport protein responsible for the efflux of cholesterol from neurones and is able to bind Aβ peptides to form stable complexes both in vitro and in vivo.11,12 Altogether, these data suggest a link between cholesterol and AD, and logically lead us to hypothesise that genes implicated in lipid metabolism may be good candidates for AD.
It is generally agreed that the best way to study complex disorders, such as AD, is by a combination of association studies and allele sharing linkage methods in pairs of affected sibs or other relative pairs. Particularly, genome scan studies provide strong evidence that at least one risk gene exists on chromosome 12.13–15 To date, three candidate genes on this chromosome have been proposed to be associated with AD. Two of them, the LRP1 and LBP1/CP2/LSF genes are located on the long arm of chromosome 12.16,17 However, only the latter has consistently shown a modest effect in independent data sets.18,19 The third gene, the α2-macroglobulin (A2M) gene, is located on the short arm of chromosome 12.20 However, the association of this gene with the risk of developing AD is still controversial and a recent meta-analysis has excluded it as a genetic determinant for sporadic AD.21
There are, however, a number of other candidate genes in this region. One of these candidate genes codes for the lectin type oxidised low density lipoprotein receptor 1 (OLR1), which is located in close proximity to A2M. Recently, a polymorphism in the 3′ untranslated region (UTR) of the OLR1 gene, +1073 C/T, has been reported to be associated with AD after stratification by APOE genotype.22 In this present study, we confirm the association of this polymorphism with AD, but independently of the APOE genotype in two different case-control samples. Furthermore, we describe a new polymorphism in the 3′-UTR, +1071 T/A, and its synergistic association with AD in conjunction with the +1073 C/T polymorphism. We also report that these two OLR1 3′-UTR polymorphisms affect the binding of nuclear proteins.
MATERIALS AND METHODS
Population description
The French AD and control samples were white (AD cases n=589, mean age = 72.3 (SD 7.2) years, mean age at onset = 69.4 (SD 7.4) years, 39.5% of men; controls n= 663, mean age = 72.5 (SD 7.9) years, 36% of men). Early age at onset was defined as ≤65 years. Diagnosis of probable AD was established according to the DSM-III-R and NINCDS-ADRDA criteria. The white controls were defined as subjects without DMS-III-R dementia criteria and with integrity of their cognitive functions. Each person or next of kin gave informed consent.
The family sample was obtained from the Indiana Alzheimer’s Disease Center (IADC) National Cell Repository that included 373 discordant sibs (230 affected, 143 unaffected) from 82 families. All affected sibs had late onset AD with mean age 74.2 (SD 6.0) years and mean age at onset 73.5 (SD 5.4) years, and 34.3% were male. Unaffected sibs had mean age 79.1 (SD 6.9) and 42.0% were male.
Brain samples
Brains were obtained at necropsy from 113 patients with early and late onset sporadic AD (mean age at onset 64.5 (SD 10.5) years, mean age at death 72.9 (SD 9.8) years, 49% were male). Genomic DNA was extracted from frozen brain tissue by standard methods and the ORL1 3′-UTR polymorphisms and the APOE genotype determined by PCR as described in the genotyping section.
The proportion of tissue area occupied by Aβ40, Aβ42, and total Aβ were quantified in immunohistochemically stained section from brodmann area 8/9 of the frontal cortex, as previously reported.23 Tau load was determined in 86 samples after immunostaining for phosphorylated Tau using a standard procedure using monoclonal antibody AT8 (Innogenetics, Belgium), as primary antibody.24 Ferritin immunostaining, as a tissue marker of activated microglia, was performed on 72 cases using a standard procedure, incubation in primary antibody (Sigma, UK) being performed overnight at 4°C at a dilution of 1:750. Sites of ferritin immunoreaction were visualised by 3,3 diaminobenzidene (DAB). Tau and microglial cell quantification was performed as described elsewhere.24 Astrocyte activation level was assessed by GFAP immunohistochemistry. Primary GFAP antibody (Sigma) was used at 1:500 and the level of staining was rated semi-quantitatively according to 0=absent, 1=few, 2=moderate, 3= high, 4=very high.24 All sections exhibited at least a few activated astrocytes and categories 1 and 2 were pooled for analyses.
Sequencing and genotyping
The 3′-UTR sequence was amplified as described by Luedecking-Zimmer et al.22 PCR products were directly sequenced using the Taq dye Terminator sequencing kit (Perkin Elmer Biosystems, Foster City, CA).
The APOE genotypes were identified by PCR followed by HhaI digestion, as slightly modified from the methods of Hixson and Vernier.26
The 3′-UTR genotypes were determined by two methods. (1) An amplification of a 156 bp fragment using mismatched reverse primer 5′-ACAAGCTAGGTGAAATAATACCG-3′ and reverse primer 5′-CTATTCTTTGTCACTTGGG-3′ was performed. The +1071 A/T genotypes were determined by AluI digestion and the +1073 C/T genotypes by MspI digestion (site created by the reverse mismatched primer) after separation on a 3% agarose gel. (2) The method described by Luedecking-Zimmer et al22 was used for the +1073 C/T genotyping using DpnII digestion while the amplicon used for sequencing was digested by AluI for the +1071 A/T genotyping. The genotyping on the French and brain samples was done in Lille, while the genotyping on the American sample was done in Pittsburgh. Approximately 10% of the genotyping performed in France was done using the two PCR methods described above and no discrepancy was observed.
Electrophoretic mobility shift assays
Cytoplasmic and nuclear extracts from lymphocyte (healthy subject) microglia cell line (CHME-5), astrocytoma (STTG-1), or neuroblastoma (Kelly) were prepared according to described methods.26 Single stranded oligonucleotides (5′-TTTTGATTCTAGCTATCTGTATTATTTCAC-3′) were end labelled with digoxigenin, annealed to complementary oligomer, and incubated for 20 minutes at room temperature with cytoplasmic or nuclear extract (5 μg). Proteins were added to a final volume of 20 ml of a mixture described elsewhere.17 The complexes were separated on a 5% non-denaturing polyacrylamide gel and semi-dry electrophoretic transfer was performed from gels to nitrocellulose membranes. Detection was as described by the supplier (Roche Diagnostics, Meylan, France).
Semi-quantitative RT-PCR assays
Purified lymphocytes from fresh blood of 20 AD cases (81.8 (SD 5.8) years, 25% of men) and 39 controls (80.3 (SD 6.5) years, 29.3% of men) were cultivated for 72 hours in the presence of 0.1% phytohaemagglutinin. Following harvesting, total RNA extraction was performed using RNeasy Mini kit (Qiagen) associated with systematic DNase treatment. Quality of the total RNA was assessed using Agilent technology indicating that the ratio of the ribosomal RNA 28S and 18S was systematically superior to 1.8. Retrotranscription was realised. From 30 ng of cDNA/RNA, real time PCR using Taqman technology was performed to coamplify cDNA from the ORL1 and β-actin genes as described by the supplier (Applied Biosystems).
Statistical analyses
The SAS software release 6.11 was used (SAS Insitute, Cary, NC). Univariate analysis was performed using Pearson’s χ2 test. In the multivariate analysis, we coded the genotype of the ORL1 polymorphisms by dichotomising genotypes into the presence or absence of a C allele for the +1073 C/T (CC+CT versus TT) and of an allele for the +1071 T/A (AA+AT/TT). The effect of these variables on the disease were assessed by a multiple logistic regression model adjusted for age and gender. Interaction between APOE and 3′-UTR polymorphisms were tested by logistic regression as well as tests for linear trends.
Pairwise linkage disequilibrium coefficients were estimated in the control samples. Extended haplotype frequencies of the two markers were estimated on collapsed data using the myriad haplotype algorithm described by McLean et al.27
Comparison of the Aβ and Tau loads, microglia and astrocyte activation, ORL1 expression according to AD status or 3′-UTR OLR1 genotypes was performed using non-parametric analyses. Results from OLR1 mRNA semi-quantification was log transformed to normalise their distributions.
RESULTS
Upon sequencing the 3′-UTR of the ORL1 gene, we found a new T/A polymorphism, two bases upstream of the previously reported +1073 C/T polymorphism (numbering from the NM_002543 sequence). We first analysed the impact of these two polymorphisms on the risk of developing AD in a French sample comprising 598 sporadic AD cases and 663 controls. The genotype distributions for both polymorphisms were in Hardy-Weinberg equilibrium (table 1). A statistically significant association with the risk of AD was observed only with the +1073 C/T polymorphism. The frequency of the +1073 C allele was significantly higher in AD cases than controls (55% v 48%, p=0.0005). Subjects bearing at least one +1073 C allele had an increased risk for AD (OR=1.56, 95% CI 1.19 to 2.04, p<0.001). No significant increased risk was observed for subjects bearing at least one +1071 A allele (OR=1.25, 95% CI 0.96 to 1.63, p=0.11). No significant interaction was detected between gender, APOE status, and the 3′-UTR polymorphisms.
Allelic and genotypic distribution of the ORL1 3′-UTR polymorphisms in the French (A) and American (B) samples
The two polymorphisms were in near complete linkage disequilibrium in the French sample (99.5%, p<0.001). Three common haplotypes, T-T, T-C, and A-C, were observed. The fourth haplotype, A-T, was observed in only three controls (0.3%) (table 2A). Two site genotypes based on three common haplotypes were assigned to each subject and haplotype frequencies compared between AD cases and controls (table 2A). The estimated haplotype distribution was significantly different between AD cases and controls. Compared to the T-T/T-T combined genotype, subjects carrying the other two haplotypes combination were at an increased risk of developing AD (table 2B). The strongest effect was observed for subjects bearing T-C/A-C, suggesting a potential but weak interaction between both polymorphisms.
(A) Haplotype distribution in the control and AD populations (B) risk of developing AD according to the haplotype genotypes
The association seen in the French sporadic AD sample was confirmed in a family based sample from the USA comprising 373 discordant sibs (230 affected, 143 unaffected). As observed in the French sample, subjects bearing at least one +1073 C allele had an increased risk for AD (OR=1.92, 95% CI 1.10 to 3.35, p=0.02) and this association was independent of APOE. Similarly, no significant increased risk was observed for subjects bearing at least one +1071 A allele (OR=1.42, 95% CI 0.81 to 2.51, p=0.22). Haplotype analysis also showed similar results in the USA sample to those seen in the French sample (table 2).
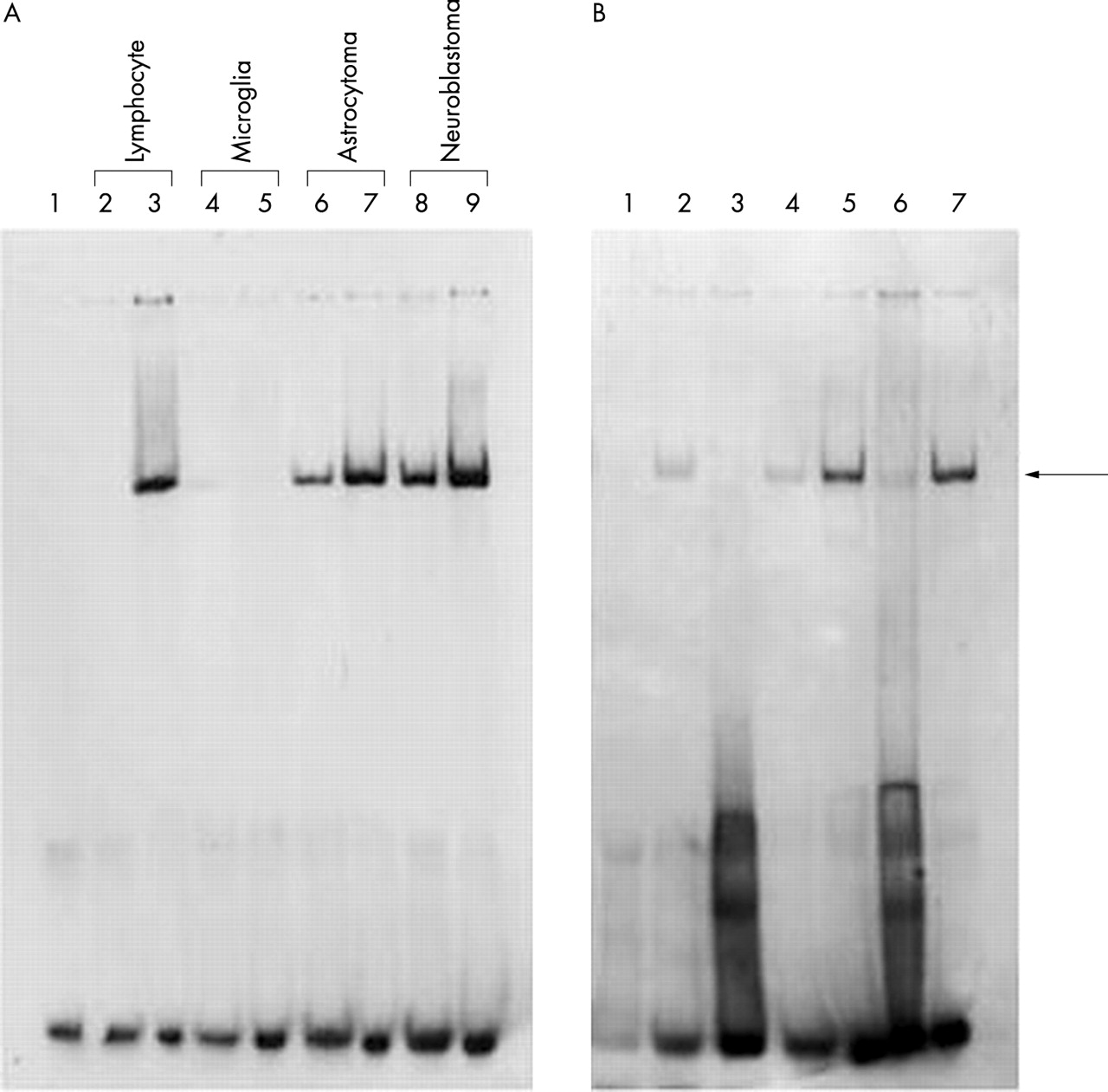
We next considered whether these two polymorphisms have biologically relevant roles consistent with modifying the risk for AD. We used electrophoretic mobility shift assays to determine if the DNA sequence spanning both polymorphisms interacts with cytoplasmic or nuclear protein extracts from different cell lines. DNA-protein complexes were observed from lymphocyte, astrocytoma, and neuroblastoma extracts, but not from the microglial cell extract. The T-T haplotype probe of the 3′-UTR OLR1 polymorphisms showed a weaker protein-DNA complex formation with cytoplasmic proteins as compared to nuclear proteins (fig 1A). The 3′-UTR OLR1 probe binding was competed by an unlabelled 3′-UTR OLR1 oligonucleotide, supporting the specificity of nuclear protein binding (fig 1B). We next compared the affinity of these proteins for oligomers containing different haplotype combinations. We found that the A-T haplotype displayed an average 1.8-fold higher affinity than the T-T haplotype under equilibrium conditions (fig 2A), suggesting that the +1071 T/A polymorphism weakly modifies the affinity of nuclear proteins for this 3′-UTR sequence. On the other hand, the T-C haplotype displayed an average six-fold higher affinity than the T-T haplotype under equilibrium conditions (fig 2B), suggesting that the +1073 C/T polymorphism strongly modulates the affinity of nuclear proteins with this 3′-UTR sequence. However, we did not detect a difference of binding affinity when comparing the T-C and A-C haplotype sequences (fig 2C). This may not be surprising, as the presence of the +1073C allele, which is associated with higher binding affinity than the +1071A allele, in both haplotypes (T-C and A-C) is probably obscuring the small affinity difference between the +1071 A and +1071 T allele. This observation is consistent with our epidemiological data suggesting at best a weak interaction between both polymorphisms.

The labelled 3′-UTR TT haplotype probes were incubated (A) with cytoplasmic (lanes 2, 4, 6, 8) or nuclear (lane 3, 5, 7, 9) protein extracts from lymphocyte, CHME-5 microglia, STTG1 astrocytoma, or Kelly neuroblastoma cell lines (negative control, lane 1) and (B) with cytoplasmic (lane 2) or nuclear proteins alone (lane 5), unlabelled 3′-UTR T-T haplotype oligo (lanes 3, 6), or scramble oligo (lanes 4, 7). Proteins were extracted from neuroblastoma (negative control, lane 1).

{kind=link}
{kind=link}
Modulation of the binding of nuclear proteins extracted from neuroblastoma by the +1071 T/A and +1073 C/T polymorphisms. Lanes show decreasing complex formation owing to increasing competition for each allele. (A) Competition of binding with increasing concentration of excess of unlabelled oligo under equilibrium conditions for the haplotypes T-T and A-T. The graph represents the relative binding of the haplotype T-T compared to the haplotype A-T. The slopes of the dashed lines that represent the initial linear phases of competition are described by the equation y=−0.059x+1.00 for the haplotypes T-T and y=−0.103x+1.00 for the haplotype A-T. (B) Competition of binding with increasing concentration of excess of unlabelled oligo under equilibrium conditions for the haplotypes T-T and T-C. The graph represents the relative binding of the haplotype T-T compared to the haplotype T-C. The slopes of the dashed lines that represent the initial linear phases of competition are described by the equation y=−0.067x+1.00 for the haplotypes T-T and y=−0.403x+1.00 for the haplotype T-C. (C) Competition of binding with increasing concentration of excess of unlabelled oligo under equilibrium conditions for the haplotypes A-C and T-C. The graph represents the relative binding of the haplotype A-C compared to the haplotype T-C. The slopes of the dashed lines that represent the initial linear phases of competition are described by the equation y=−0.381x+1.00 for the haplotypes A-C and y=−0.393x+1.00 for the haplotype T-C.
In order to characterise better the potential impact of these polymorphisms on the aetiology of the disease, we examined the effect of these two polymorphisms on Aβ and Tau loads and astrocyte and microglial cell activation in AD brains. No impact of this polymorphism was observed on Aβ, Tau loads, or microglial cell activation (data not shown) but a possible association of the +1073 C/T polymorphism with astrocyte activation was observed, the +1073 C allele being associated with a higher level of activation (table 3). The low frequency of the +1071 T/A polymorphism did not allow meaningful statistical analysis (data not shown).
Level of astrocyte activation according to the ORL1 +1073 C/T polymorphism in brains of AD cases
We finally investigated whether these differences were reflected by altered expression of the OLR1 gene. Using a semi-quantitative RT-PCR assay, we estimated the ratio between the amount of the OLR1 and β-actin mRNA from lymphocytes of control, AD, and young subjects. The level of expression of the OLR1 gene in relation to the β-actin gene was significantly lower in AD cases compared with controls (p=0.009) (table 4) and this difference was confined to subjects bearing the CC and CT genotypes (p=0.009). These data indicate that the +1073 C allele may be functional by lowering the ORL1 expression in AD lymphocytes more than controls. The low frequency of the +1071 T/A polymorphism did not allow meaningful statistical analysis (table 4).
Level of OLR1 mRNA in lymphocytes from AD cases and elderly controls according to the 3′-UTR polymorphisms
DISCUSSION
The 3′-UTR +1073 C/T polymorphism has been shown to modify the risk of sporadic AD in an APOE dependent fashion.22 In this paper, we report the association of the 3′-UTR +1073 C/T polymorphism with the risk of AD independent of APOE status in two different samples comprising French sporadic and American familial AD cases. We also describe a new polymorphism, +1071 T/A, located two bases upstream of the +1073 C/T polymorphism. Although the latter polymorphism was not associated with AD by itself, the two site haplotype analysis suggested a potential weak combined effect of these two polymorphisms on the risk of AD.
The relevance of the OLR1 3′-UTR polymorphisms, especially the +1073 C/T polymorphism, to the risk of AD is highlighted by the functional experiments we performed. Indeed, we show that the 3′-UTR sequence encompassing the two polymorphisms binds differentially to regulatory proteins; the +1071 A and +1073 C alleles bind with higher affinity compared to the +1071 T and +1073 T alleles, respectively. Furthermore, the highest level of astrocyte activation in AD brains may be associated with the presence of the +1073 C allele. Finally, the +1073 C allele may be associated with a specific decrease of OLR1 expression in lymphocytes from AD cases.
In addition to these positive associations, there are, however, some caveats that need to be considered since no effects of the 3′-UTR polymorphisms were observed on Aβ and Tau loads. Similarly, microglial cell activation seems not to be modulated by these polymorphisms. However, this latter observation might be expected, as no DNA-protein complex was observed using protein extract from a microglial cell line. On the other hand, the 3′-UTR sequence formed a DNA-protein complex with the astrocytic proteins and the astrocytic activation was affected by the +1073 C/T polymorphism. These data suggest that OLR1 may act through a specific local mechanism involving astrocytes.
Another important point is the significant reduction of OLR1 expression in lymphocytes (−362%) in AD cases compared to controls indicating that control of the expression of this gene is potentially important for the development of AD. However, a quantification from brain tissue appears necessary since the expression of OLR1 is about five-fold higher in different parts of the brain than in peripheral lymphocytes.28 Furthermore, we cannot exclude the possibility that the potential effect of the 3′-UTR polymorphisms on OLR1 expression is specific to brain tissue, even restricted to one cell type, such as the astrocyte.
The exact mechanism by which OLR1 may affect the risk of AD is not clear as yet. OLR1 is abundantly expressed in the human central nervous system (CNS),28 but its physiological role has not been evaluated. Despite the existence of the blood-brain barrier, lipoprotein particles have been shown also to be present in the cerebrospinal fluid (CSF). Although some of these protein components may filter through the barrier from the vascular compartment, experimental evidence indicates that these particles originate from nervous tissue.29 Furthermore, it has been shown that lipoprotein particles can be isolated from the media of astrocytic cultures.30 CSF and CNS lipoproteins seem to be vulnerable to oxidative modifications.31 This is of particular relevance to Alzheimer’s disease since reactive oxygen species are abundantly produced following chronic inflammation.32 Lipoprotein oxidation may disrupt several mechanisms leading or facilitating the disease. (1) Oxidised LDL (oxLDL) or oxidised HDL (oxHDL) may directly induce neuronal death in vitro. Coapplication of oxLDL or oxHDL with amyloid peptide, which enhances oxidative stress, resulted in increased neuronal death.33,34 (2) Aβ peptides are associated with lipoproteins secreted by cultured astrocytes. We may suppose oxidation of these lipoproteins may modify their ability to bind amyloid fibrils, disrupting a potential mechanism of Aβ clearance.35 (3) The growth and maintenance of an elaborate neurite network requires directional lipid transport; alterations in this lipid transport might be detrimental to neuronal integrity. It has been proposed that neurones must import glial cell derived cholesterol via lipoproteins to form numerous and efficient synaptic connections.36 In this way, the main phase of synaptogenesis starts synchronously after glial cell differentiation throughout the CNS.37 Neurological symptoms of AD may be associated with defective cholesterol and lipoprotein metabolism.6 Disruption of this metabolism may directly result from lipoprotein oxidation or from interaction with Aβ peptides. Indeed, it has recently been suggested that Aβ peptides may directly regulate cholesterol transport and homeostasis via lipoproteins and overproduction of Aβ or high cholesterol may block cholesterol trafficking.38 Oxidation of lipoproteins may accentuate this mechanism.
In addition to being functionally relevant to affecting the risk of AD, OLR1 is also a positional candidate gene for AD based on the linkage mapping data in white13–15,39,40 and non-white41 families. Indeed, a repeat polymorphism in the 3′-UTR of OLR1 has shown the strongest evidence of linkage on chromosome 12 with AD.40 Recently, although Blacker et al42 have reported the absence of linkage on chromosome 12 in the full NIMH AD Genetics Initiative families (411 white, 26 non-white), they continue to see strong association with A2M, which is located in close proximity to OLR1 on chromosome 12. Furthermore, significant evidence of linkage on chromosome 12 has been reported in 230 of the NIMH families,39,40 Thus, our current OLR1 association data in conjunction with earlier OLR1 association22 and linkage13–15,39–41 data strongly suggest that the OLR1 gene may directly affect the risk of AD. This study, however, does not exclude the possibility that the effect observed here could be attributable to linkage disequilibrium with a functional mutation in the promoter or 5′-UTR region of OLR1 or to linkage disequilibrium with functional mutations in nearby genes. Additional analyses of the OLR1 gene or nearby genes may help to identify the chromosome 12 AD gene.
In conclusion, our data suggest that the OLR1 gene may be a risk factor for AD. This observation, combined with other studies showing that other genes coding for the LDL receptor family may be genetic determinants of AD, underlines the importance of cholesterol trafficking in the aetiology of AD.