Article Text

Abstract
MSX1 has been proposed as a gene in which mutations may contribute to non-syndromic forms of cleft lip and/or cleft palate. Support for this comes from human linkage and linkage disequilibrium studies, chromosomal deletions resulting in haploinsufficiency, a large family with a stop codon mutation that includes clefting as a phenotype, and the Msx1 phenotype in a knockout mouse. This report describes a population based scan for mutations encompassing the sense and antisense transcribed sequence of MSX1 (two exons, one intron). We compare the completed genomic sequence of MSX1 to the mouse Msx1 sequence to identify non-coding homology regions, and sequence highly conserved elements. The samples studied were drawn from a panethnic collection including people of European, Asian, and native South American ancestry. The gene was sequenced in 917 people and potentially aetiological mutations were identified in 16. These included missense mutations in conserved amino acids and point mutations in conserved regions not identified in any of 500 controls sequenced. Five different missense mutations in seven unrelated subjects with clefting are described. Evolutionary sequence comparisons of all known Msx1 orthologues placed the amino acid substitutions in context. Four rare mutations were found in non-coding regions that are highly conserved and disrupt probable regulatory regions. In addition, a panel of 18 population specific polymorphic variants were identified that will be useful in future haplotype analyses of MSX1. MSX1 mutations are found in 2% of cases of clefting and should be considered for genetic counselling implications, particularly in those families in which autosomal dominant inheritance patterns or dental anomalies appear to be cosegregating with the clefting phenotype.
- MSX1
- birth defects
- cleft lip and palate
- homeobox
Statistics from Altmetric.com
Non-syndromic cleft lip with or without cleft palate (CL/P) and cleft palate only (CPO) occurs with wide geographical distribution with a range of birth prevalences from high in Asians and native Americans to intermediate in European and low in African populations.1 Numerous models have been suggested to best explain the inheritance of CL/P and CPO since the initial genetic descriptions of Fogh-Anderson2 and Fraser.3 Gene-gene and gene-environment interactions almost certainly play a role4 and identification of specific influences has expanded in recent years with the availability of molecular and epidemiological studies. Several loci and genes have been suggested as candidates.5,6 Candidate gene studies have been especially productive in non-syndromic clefting since the original description by Ardinger et al7 of an association with alleles at TGFA. In recent years MSX1 is emerging as an especially strong candidate based on the cleft palate and foreshortened maxilla phenotype in the mouse knockout.8 Association studies of MSX1 with CL/P and CPO9–14 further support a role for MSX1 in non-syndromic clefting in different populations. A report in a family with a nonsense mutation at position 105 in MSX1 segregating in an autosomal dominant fashion for both clefting and tooth agenesis15 has suggested that, particularly in familial cases, MSX1 mutations might be identified. To determine whether mutations in MSX1 might be aetiological in a substantial portion of non-syndromic cleft lip and palate cases, we undertook a complete sequencing effort to determine the prevalence of mutations in DNA from a large selection of CL/P and CPO cases born in multiple geographical regions.
METHODS
Study populations
All samples were collected with signed consent and had local and/or University of Iowa IRB approval. Samples from outside the USA were anonymised.
Samples from South American countries were collected through the Latin-American Collaborative Study of Congenital Malformations (ECLAMC).16 From January 1998 to June 2000, ECLAMC collected blood spots on filter cards from 233 patients with cleft lip with or without cleft palate (CL/P) and cleft palate only (CPO) and their mothers from eight countries in South America: Argentina, Brazil, Bolivia, Chile, Ecuador, Paraguay, Uruguay, and Venezuela.
Cases from the Philippines were studied under the auspices of Operation Smile International.17 Samples were from 296 CL/P and CPO patients who were seen and examined at one of four sites within the Philippines (Cavite, Kalibo, Cebu, and Negros).
Blood spots on filter cards were collected through the Japanese Cleft Palate Foundation (Aichi-Gakuin University, Nagoya, Japan), which is a medical organisation that provides clinical care for underserved populations in Japan, Vietnam, Mongolia, and Cambodia.18 Samples were from 41 CL/P and CPO patients who were seen and examined in 1996 and 1997 at Nagoya, Japan, and Bentre area, Vietnam.
Cheek swabs from 147 triads (case, father, mother) were collected between October 1999 and March 2000 according to a standard protocol. Volunteers were CL/P and CPO families contacted through the University of Southern Denmark.
Blood specimens were collected through the Iowa Birth Defects Registry.19 Samples were from 200 CL/P and CPO available children and their parents, who were seen and examined between 1 January 1987 and 31 December 1991.
Controls comprising unaffected subjects were obtained from different populations. DNA samples from 37 unrelated Brazilians who were unaffected or had a child with Van der Woude syndrome (OMIM Accession No 119300) or mandibulofacial dysostosis syndrome (OMIM Accession No 604830), and DNA from 20 Native Indians from the Coriell Cell Repositories (14 from the Rondonia Province, Brazil, four from South Central Andes, Peru, one from Campeche State of the Yucatan, Mexico, and one from Ecuador) comprised the controls from South America.
A control population from Asia20 comprised a sample of 544 consecutively born newborns in Bacolod City, Philippines. Cheek swab samples from 142 subjects from Negros, Philippines, collected in February 1994, and 30 samples from the Coriell Cell Repositories (10 from Japan, 10 from China, and 10 from South Asia) completed the controls from Asia. One hundred Iowa controls were selected from a group of all live births between 1 January 1987 and 31 December 1991 without a birth defect using the Iowa Birth Defects Registry.20 An additional 170 samples of DNA from unrelated whites were obtained from CEPH (Centre d’Etudes du Polymorphisme Humaine) families.21
PCR primers
Primers were prepared and used to amplify overlapping regions of the MSX1 gene (Electronic appendix 1: http://genetics.uiowa.edu/publications/peterj/PeterMSX190902.html, fig 1). The regions examined were: exon 1, exon 2, and the intron from 468 nucleotides 5‘ of the start codon, including the complete 5‘ untranslated region (UTR), to 1073 nucleotides past the stop codon (including the complete 3‘ UTR). This region included a 389 nucleotide section of the intron (with 80% nucleotide homology to the mouse sequence S73812) and all of the antisense transcript in MSX1.22
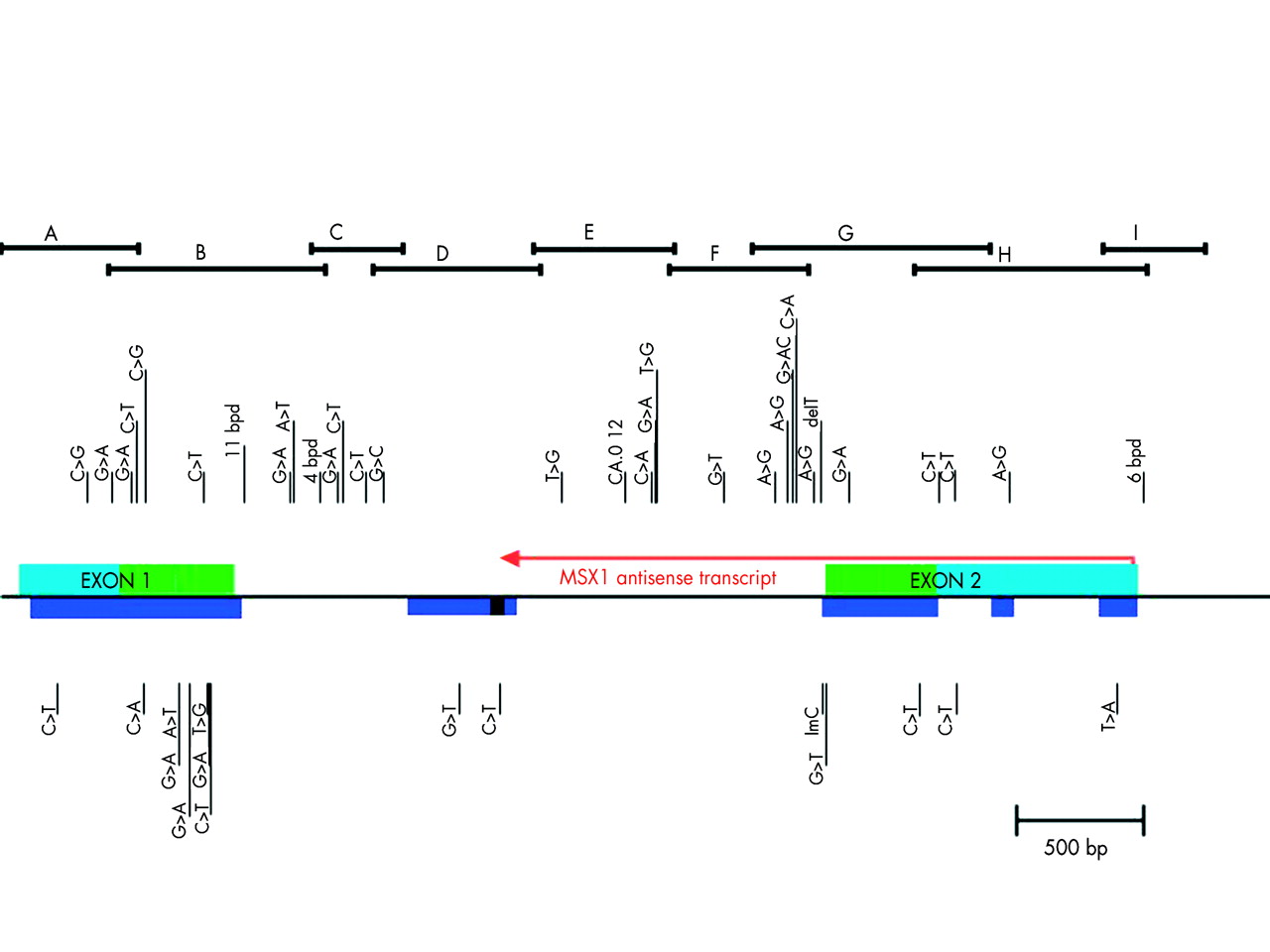
Structure of the MSX1 gene and description of the variants found. Horizontal bracket bars on top indicate overlapping PCR products sequenced as described in text. Variant sites are shown by vertical lines with the letters of the nucleotide change indicated. Common alleles are indicated above and potential aetiological mutations are indicated below the gene structure. Light blue indicates 5‘ and 3‘ UTR. Green indicates the coding region of exons. Dark blue indicates the regions of human/mouse sequence homology. Red arrow represents the antisense-MSX1 transcript length/direction. Black box within the dark blue is the range where antisense transcript terminates.
DNA extraction and amplification
DNA was extracted from filter card blood spots using modifications of published protocols20,23,24 and from cheek swabs25 and whole blood26,27 according to published protocols. PCRs were performed in 25-50 μl volumes containing 10-20 ng DNA/μl; 200 μmol/l each of dATP, dCTP, dGTP, and dTTP; 1.5 mmol/l MgCl2; 10 mmol/l Tris/HCl pH 8.3; 50 mmol/l KCl; 0.001% (w/v) gelatin; 0.25-1.0 μmol/l of each primer; and 0.01-0.02 unit Taq polymerase/μl. Following assembly, the reactions were covered with 50 μl of mineral oil.
DNA sequencing
Following DNA amplification, unincorporated PCR primers and deoxynucleotide triphosphates in the sample were removed before sequencing by isolation of the desired band in a 2% agarose gel coupled with column purification.28 Cycle sequencing was performed in a 20 μl reaction using 4 μl of ABI Big Dye Terminator sequencing reagent (version 2 or version 3), 1 μl of 5 μmol/l sequencing primer, 1 μl DMSO, 4 μl of 2.5 × buffer, and 2.5 ng/100 bp of DNA template. Following a denaturation step at 96°C for 30 seconds, reactions were cycle sequenced at 96°C for 10 seconds, 50°C for five seconds, and 60°C for four minutes for 40 cycles. Cleanup of version 2 chemistries was performed using Centricep spin columns (Princeton Separations, Adelphia, NJ). The products were evaporated to dryness under pressure (Savant Instruments, Farmingdale, NY), resuspended in 2-4 μl loading buffer, heated for four minutes at 95°C, and 1 μl loaded on an Applied Biosystems 373SL sequencer. Cleanup of version 3 chemistries was performed on a Multiscreen Sequence Plate, 384 well format (Millipore, Bedford, MA), resuspended in 40 μl, and 2.5 μl injected on an Applied Biosystems 3700 sequencer.
Sequence analysis
The ABI sequence software (version 2.1.2) was used for lane tracking and first pass base calling (Perkin Elmer). Chromatograms were transferred to a Unix workstation (Sun Microsystems Inc, Mountain View, CA), base called with PHRED (version 0.961028), assembled with PHRAP (version 0.960731), scanned by POLYPHRED (version 0.970312), and the results viewed with the CONSED program (version 4.0).29 When the results indicated a possible new variant, the sample was resequenced with available parents or other family members, and the new sequences were analysed using the same method.
Statistical analysis
Standard chi-square and Fisher’s exact probabilities were evaluated on all sets of comparisons. Significance figures were accounted for using Bonferroni correction taking into account the number of tests carried out. With the Bonferroni correction, alpha will be 0.0009 (0.05/53 comparisons). Linkage disequilibrium was calculated using D’ statistics using the software package GOLD (Graphical Overview of Linkage Disequilibrium).30 Values higher than 0.9 are considered in strong linkage disequilibrium and a value equal to 1.0 indicates complete linkage disequilibrium.31
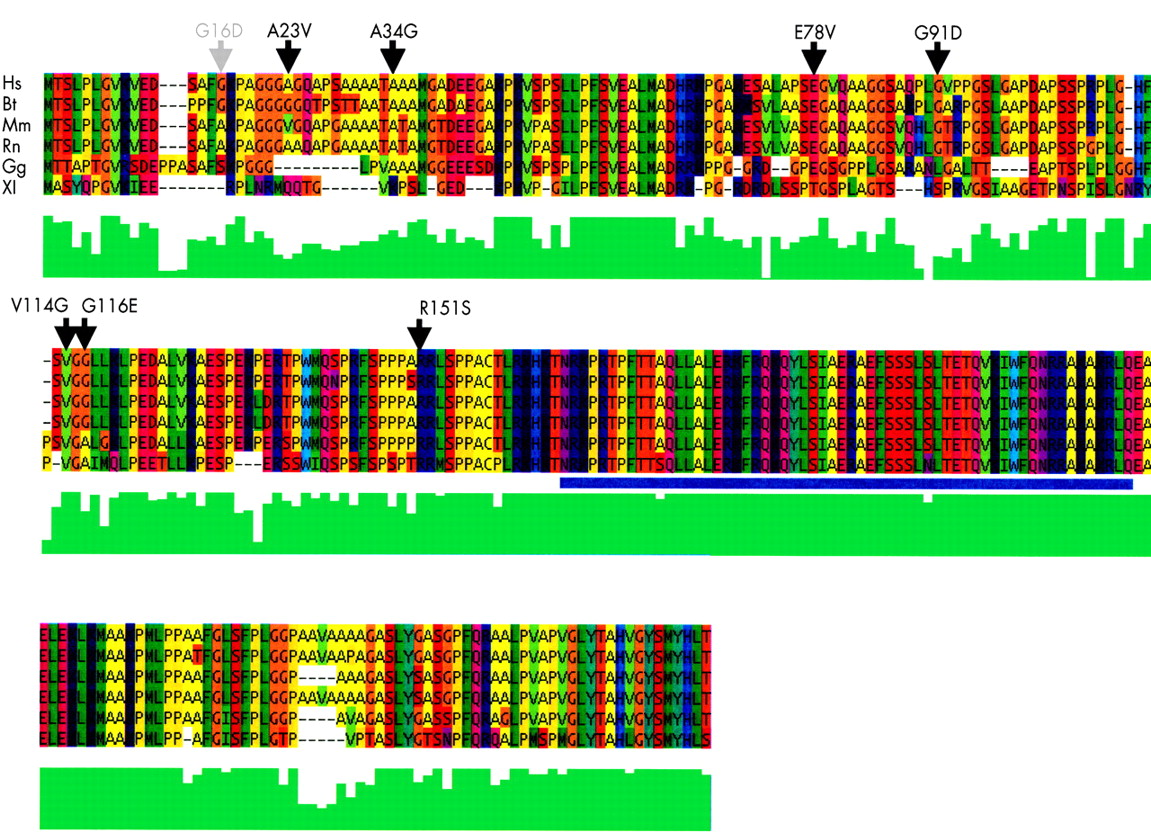
Protein sequence comparisons
Msx1 orthologues were first identified through a BLAST search of the non-redundant database using Homo sapiens MSX1, accession NP_002439.1, as the reference sequence. All known and complete Msx1 sequences were included from the vertebrate lineage. These files in FASTA format were then manipulated in Jalview (Jalview: http://www2.ebi.ac.uk/∼michele/jalview/contents.html) and submitted for remote alignment at the EBI using a Clustal algorithm. Minor adjustments were made in the Jalview editing program to correct only obvious discrepancies in sequence alignments. The human MSX1 sequence is aligned with the cow (Bt 302960), mouse (Mm 123311), rat (Rn 13592001), chick (Gg 1708273), and clawed toad (Xl 547690) sequence using remote Clustal alignment from EBI. The multiple protein sequence alignment of the Msx1 protein from different species shown in fig 2 is coloured with the Taylor colour scheme used within Jalview. In this scheme, the colours are set by the variation in polarity and size of different amino acid chains. Small hydrophobic amino acids are coloured green, large aromatic amino acids are green/blue, large polar/basic amino acids are purple/blue, and small polar/acidic amino acids are orange/red. Jalview provides a visual assessment of the degree of amino acid consensus at any site as per the ClustalW algorithm. The ClustalW amino acid homology quality score provided from within the Jalview program is represented by the vertical green bars under the sequence alignment of the MSX1 putative orthologues. This qualitative quality score makes it possible to estimate the significance of a given amino acid substitution within the alignment. It measures both amino acid class conservation as well as evolutionary conservation at any given site.

Protein sequence alignment for MSX1. The human MSX1 sequence is aligned with the B taurus, M musculus, R norvegicus, G gallus, and X laevis sequence using ClustalW. Non-conserved amino acid variants from controls (grey) and cases (black) are indicated with arrows above the appropriate residue. The green bar graph below each position is a quantitative ClustalW function to show the degree of sequence homology. The homeobox region is underlined by a blue bar.
RESULTS
Genomic structure of MSX1
The single 2 kb intron of MSX1 was initially amplified with one primer pair set and sequenced using primer walking. A schematic model of the gene structure and sequence can be seen in fig 1, and sequence data are available via GENBANK accession number AF426432. The sequence of the MSX1 gene was then compared to human sequences available via GENBANK. This included comparisons to MSX1 cDNA, BLAST matches from the EST databases, and genomic contigs of this region of chromosome 4 (accession numbers M76731, AH002826, M76732 NM_002448, XM_003469 and NT_006148).32,33 Similar comparisons were made to the Msx1 sequence of mouse (S73812, NM_010835) and other species by BLAST to determine regions of homology. Those cross species homology regions with greater than 78% nucleotide identity over at least 50 bp are indicated in fig 1 by solid colour underbars. In addition, Blin-Wakkach et al22 identified the presence of an antisense transcript that overlaps the intron, 3‘UTR, and all of exon 2. It is also indicated in fig 1. It appears to terminate within the MIHR (MSX1 Intronic Human-mouse Homology Region) identified by our BLAST comparison between our sequence and the genomic mouse sequence. A 98 amino acid open reading frame of unknown significance is found within the antisense transcript just before its termination in the MIHR.
Polymorphism and mutation identification
Nomenclature herein follows those recommendations at the Human Gene Mutation Database (http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html). For the sake of brevity, the position of variants may be referred to by the nucleotide position within the Genbank entry, AF426432. A summary of the MSX1 mutations and polymorphisms found in this study is available electronically at: Electronic appendix 2, http://genetics.uiowa.edu/publications/peterj/PeterMSX190902.html. Several variants were described previously.10,20,33,34 A total of 917 DNA samples from subjects with non-syndromic cleft lip and palate and cleft palate only (799 CL/P, 118 CPO) and 1027 DNA samples from unaffected subjects were screened in this study. Only 15 cases had an associated anomaly besides clefts. In addition, 324 parents were screened in order to validate the new variants found.
This sequencing effort concentrated on the putative functionally important regions of this locus as defined bioinformatically. Thus the coding regions and a large conserved mouse sequence homology element in the single intron received priority. In addition, the remainder of the locus from putative sense to antisense promoter were also screened in a minimum of 100 cases to identify both rare and common sequence variants in these regions. The sense promoter was defined by sequence homology to the functionally defined murine MSX1 sense minimal promoter.35 The antisense (AS) promoter region has murine sequence homology upstream of the AS transcript’s start site that includes CAAT and TATA boxes.
From this approach, 48 variant sites that included three small deletions (11, six, and four nucleotide deletions, all in non-coding regions), the previously reported CA repeat in the intron, one common single base deletion, one rare single base insertion, and 43 single nucleotide polymorphisms (SNPs) were identified and mapped. The 43 SNPs included one site with two SNPs (3118 G>A,C). These 43 SNPs comprised 27 transitions (seven in coding regions) and 16 transversions (five in coding regions). There was a preponderance of C>T transitions (13) over all other changes noted and five of these were initially part of CpG dinucleotides. We defined a sequence variant as common if it was found in more than 1% of any population in this study.
Non-coding variants
In the 5‘UTR four SNPs were found, all at sites of sequence conservation between mouse and human (as determined by comparisons to the mouse sequence at the UCSC golden path database, the Mouse Sequencing Consortium via: http://genome.ucsc.edu/). The –247C>T variant was found in a syndromic cleft case from Iowa in a highly conserved sequence block homologous to the mouse Msx1 minimal promoter.35 The proband has multiple congenital anomalies in addition to bilateral cleft lip and cleft of the hard and soft palate. These include choanal atresia, anophthalmia and microphthalmia, congenital cataracts, pachygyria, developmental delay and/or mental retardation, and dipigmentation. The pigmentation is a form of skin mosaicism mixing pigmented and non-pigmented areas in the body. DNA samples from tissue biopsies of both pigmented and non-pigmented skin patches were both heterozygous for the C/T variant. The proband’s mother has a brother with non-syndromic bilateral cleft lip and palate. A second variant (−167C>T) was found in a white (CEPH) control, and it is located in the 21 bp segment that is duplicated in mouse but not in human. The clefting status of this control case is unknown. The other two SNPs in the 5‘UTR were both found at high frequencies in control populations.
The end of the sense transcript colocalises with the start of the antisense transcript. In the sense 3‘UTR, five more SNPs were found. To help map the end of the sense 3‘UTR, BLAST was performed between AF426432 and the human EST database. This showed several transcripts whose 3‘ ends extend close to the position of the 3‘ region of homology between the human genomic sequence and the mouse mRNA sequence, thereby establishing a presumed 3‘UTR terminus for the MSX1 gene. The mouse mRNA/human genomic homology extends to position 4469 of the AF426432 sequence. The homology between the most 3‘ human EST fragment (AA464792) and AF426432 extends to position 4477 in AF426432. This presumed termination of the MSX1 transcript differs substantially from the position mapped previously at position 4270.33 There is an AATAAA, poly A signal at 4449, just upstream of this termination site in both human and mouse. If this is in fact the real termination of the MSX1 transcript, it is remarkably close to the start of the antisense MSX1 transcript, at position 4396, as mapped by Blin-Wakkach et al.22
One sense 3‘UTR variant at position 3969 was found commonly and within a conserved mouse sequence block. A different sequence variant, a 6 bp deletion was found 22 nucleotides beyond the end of the sense transcript and just 104 nucleotides before the start of the antisense transcript. The 6 bp deletion is found eight nucleotides upstream of the CAAT box for the antisense transcript. The CAAT box itself was previously identified22 to be 32 nucleotides upstream of an extremely conserved sequence element that contains the TATA box and the transcription start site for the antisense transcript. The 6 bp deleted sequence element itself is partially conserved (five of six nt) in comparison to the aligned mouse genomic sequence from this region. A third SNP, found at position 4401, is within this highly conserved sequence element (conserved back to Fugu by NCBI-BLAST, data not shown). SNP 4401 is five nucleotides upstream of the antisense transcription start site in human sequence and 24 nucleotides downstream of the TATA box. This conserved element is shown at the far right in fig 1 by the solid colour underbar at the start of the antisense transcript.
Two different point mutations were found in the MSX1 intronic homology region (MIHR) in two Iowa probands, but no other changes within the MIHR were found among the other approximately 900 subjects. The MIHR region overlaps the termination of the MSX1 antisense transcript.22 One of the two variants (451+887G>T) was found between the location of two antisense primers used to identify the MSX1 antisense transcript termination. However, this nucleotide position was not conserved in the mouse sequence of this region, although it is conserved across primates (data not shown). The other SNP identified in the MIHR was found at a site of conservation with the mouse sequence (451+1046C>T).
Protein sequence alignment
Fig 2 shows the amino acid sequence for humans MSX1 aligned to the sequence from the cow, rat, mouse, chicken, and frog. Amino acid variants discovered in the course of this study are indicated by vertical short arrow heads. The colouring of amino acids by their chemical class helps to visualise such differences in an evolutionary context. The coding regions of MSX1 contained 14 different SNPs, six synonymous and eight non-synonymous, with exon 1 bearing the majority of coding SNPs (11).
Coding variants
The amino acid change that occurred in the most conserved position was the R151S change. The arginine in this position is completely conserved back to the axolotl as a double R motif and a single R is conserved back to Drosophila. The R151S change is found in the conserved block of amino acids just N-terminal to the homeodomain, previously called the extended homeodomain (EHD).36 This EHD region includes an apparent PBX binding motif (TPWMQ37), several potential phosphorylation sites, a potential nuclear localisation signal, and conserved residues that mediate homo- and heterodimerisation of MSX1 with other transcription factors.38 The other amino acid variants have varying degrees of evolutionary sequence similarity. The V114G change disrupts considerable evolutionary conservation, in this case back to X laevis. The V114G variant is found in a family with an unaffected sib who carries the same mutation inherited from the unaffected (no cleft) mother.
In the case from Uruguay, two variants were found only eight nucleotides apart. The first one is a missense mutation G347A (G116E) and the other one is a nucleotide change that does not alter the amino acid sequence (C354T, L118L). This subject has bilateral cleft lip with cleft palate and a dizygotic twin who has unilateral cleft lip with cleft palate, but neither of these two variants. One could argue that the missense mutation, or a combination of the two variants together might increase the severity of the defect or be acting in diallelic inheritance.39,40
Synonymous coding variants
As previously mentioned, the coding portion of exon 2 was remarkable for the lack of sequence variation when compared to exon 1. Exon 2 exhibited only three different SNPs/443 nt (0.68%) while exon 1 had 11 different SNPs/451 bp (2.4%) in all the samples (p=0.04). This conservation suggests the presence of hidden regulatory elements, preferentially in exon 2. The single variant at position 3620 is found within the coding portion of exon 2. It is a synonymous nucleotide change in alanine 275. However, this change may affect the splicing process as it eliminates a significant ESE (exon splice enhancer) at this site. The ESE finder at http://exon.cshl.org/ESE/ predicts a SRp55 ESE with a score of 2.70. This ESE is disrupted in the presence of the A275A, 825 C>T mutation. It has been shown that even constitutively spliced exons, like MSX1 exon 2, require the presence of splice enhancers,41 presumably to ensure correct splicing. Notably, a potential pseudo splice acceptor (rated 0.98 by the human splice junction algorithm at http://www.fruitfly.org), is found in the intron at position 2566-2606 close to the correct exon 2 junction at 3226-3266 (which was rated as 0.97 by the same algorithm). The sum of all ESEs in exon 2 may be required to ensure correct splicing and protect against potential aberrant splicing.
Table 1 shows all of the rare variants, both coding and non-coding, identified in this study that are potential disease causing mutations. Fifteen variants are listed in a total of 16 subjects. Only one of these variants (A233T, see below) was identified in 500 controls sequenced. Five variants are missense mutations. An additional missense mutation was found in two Brazilian controls at position G47A (G16D). The missense mutation A233T (E78V) was found in four Filipino cases (two related subjects) and in one control from the same population. The clinical status for clefting of the control is unknown as it came from a collection of 500 consecutive anonymised newborns where the frequency of clefts is 1/500.42 All cases with the A233T variant have a positive family history for clefts with the mutation found in two other affected relatives available for study.
Potential mutations found in the present study (total number of cases screened by population Asians, whites, and South Americans for each site is described between parentheses)
Given the case and control numbers shown in Electronic appendix 2, a power calculation suggests that anywhere from 600 to 12 000 controls would have to be sequenced to show statistical significance of any one of these rare variants with the disease, given that no more cases would be sequenced and the proportions of cases and controls remained the same. This is not practical so other measures can be used to assess their relevance. We grouped all variants presented in table 1 and compared them to controls. A p value = 0.017 is calculated for the comparison of the disease associated variants against the absence of those same variants in the controls. If we tested each of the 15 variants with no Bonferroni correction, an alpha for each test would be 0.003 for significance. So by a conservative test no single change is significant. Further, these rare variants are found in phylogenetically conserved amino acid or genomic sequence.
Common variants
From the set of all the variants, 26 are present as common alleles. From this subset, 18 of these show marked population differences that may facilitate future population specific haplotype-disease association studies. Among all these population specific alleles, two were found to have significant association (table 2). At position 799, 330 C>T, G110G, a synonymous coding SNP was found significantly associated with the Asian case population (p=0.03, no Bonferroni correction). This site was previously shown to be significantly associated with cases in an Iowan population,10 but does not replicate in the larger number of Iowa cases reported here. For the variant at position 4500, *(811-816)del, an association within the Iowan population was observed (p=0.03, no Bonferroni correction).
Case-control comparison of population specific alleles
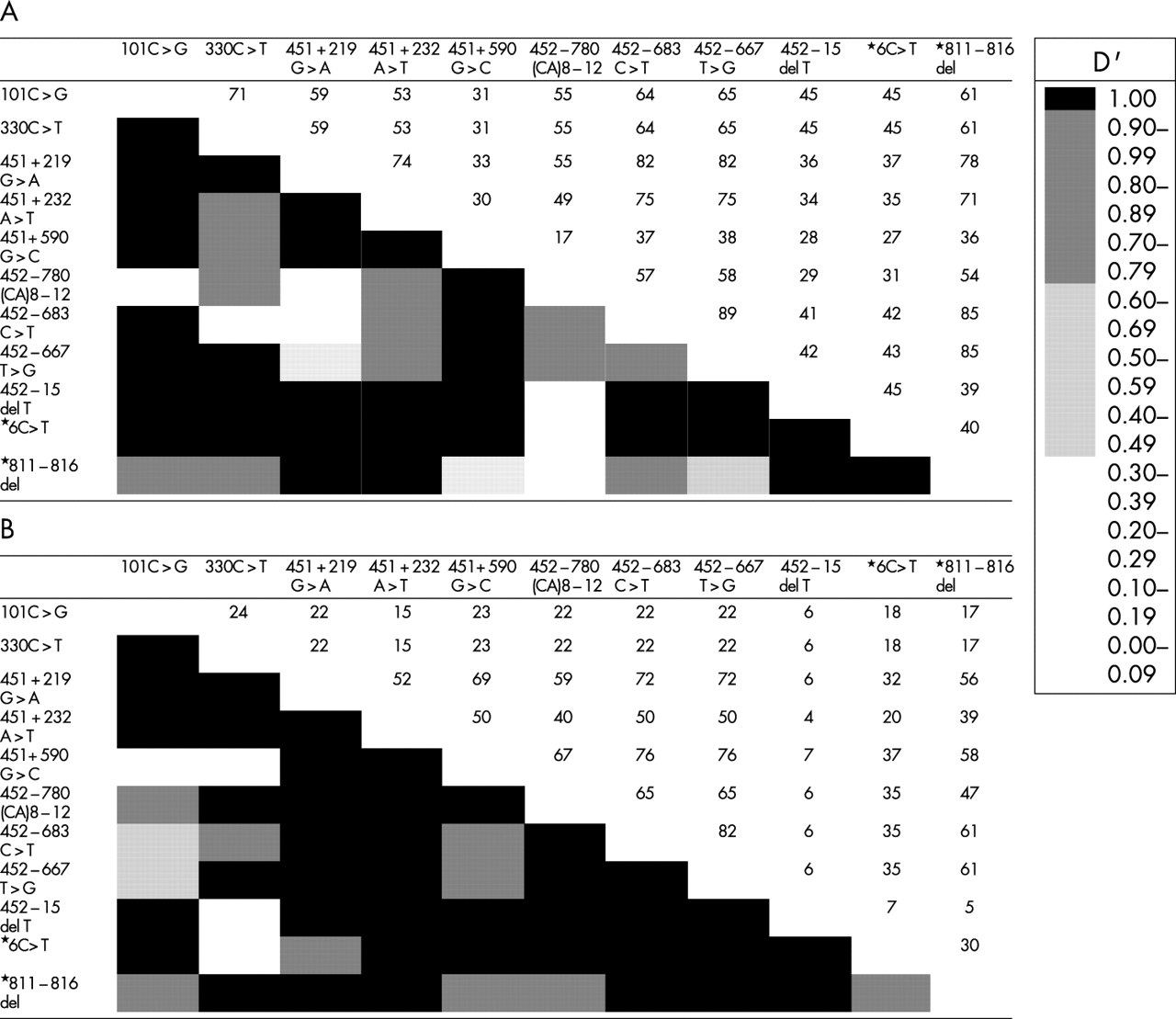
Fig 3 shows linkage disequilibrium for 11 sites with allelic variants with heterozygosity higher than 0.05 in controls. Polymorphic sites across the MSX1 locus were selected according to their allele frequency. Presence of linkage disequilibrium between two random sites is expressed by D’ values higher than 0.5. The MSX1 CA4 repeat was most often found in linkage disequilibrium to the less common allele at each variant site (data not shown). It is remarkable that a cluster of polymorphisms is found around the variant 452-780 (CA)8-12, especially in the Iowa population, where previous studies have indicated a disease association with the CA4 repeat marker. Haplotype analysis can now be done using selected variants.

{kind=link}
{kind=link}
{kind=link}
Linkage disequilibrium analysis of the MSX1 locus for (A) the Filipino population and (B) the Iowa population. Different shaded blocks represent D’ statistics and data represented as numerals are chromosome numbers.
DISCUSSION
Common birth defects such as neural tube defects, congenital heart disease, and cleft lip and palate can occur in both syndromic (which include structural abnormalities, developmental delay, or dysmorphic features), and non-syndromic forms. While both genes and environment play a role in the non-syndromic forms, the specifics of these have remained largely undiscovered until recently. For neural tube defects, a strong environmental component is clearly shown by the ability of prenatal folic acid to prevent a proportion of these cases.43,44 Genetic factors, perhaps including predispositions to the effects of folate or relative folate deficiency, seem likely also to play a role. For congenital heart disease, endophenotypes such as tetralogy of Fallot in apparent non-syndromic forms has been reported associated with NKX2.5 mutations, which suggests a specific gene component to this formerly complex trait in 4% of families.45 Thus, searches for mutations in single genes whose defects include CL/P or CPO may also disclose specific abnormalities in non-syndromic forms. Examples of genes causing syndromic forms of clefting in which non-syndromic forms might be masked include the Van der Woude syndrome and the IRF6 gene when lip pits are absent,46 EEC syndrome and P63 gene mutations,47 Margarita Island ectodermal dysplasia syndrome,48 X linked ankyloglossia and cleft palate,49 and the MSX1 gene in which clefting,15 selective hypodontia,50 or dental anomalies associated with other ectodermal features51 are found with specific coding sequence mutations.
Samples included in this study were comprised primarily of subjects who have isolated cleft lip as well as cleft lip and palate. Although historically cleft palate only has been separated aetiologically and embryologically from clefts involving the lip or the lip and the anterior hard palate, recent work suggests that this may not be an absolute division. Families such as that of van den Boogaard et al,15 as well as published and anecdotal observations of clinical cases suggests that isolated cleft palate can occasionally occur in the context of cleft lip and palate.52 Similarly, the linkage disequilibrium data have, in some cases, supported a role for both cleft palate and cleft lip and palate together.10 One recent study suggests that the important distinction is not whether the lip is involved but whether the palate is included.13 It may be important to investigate both lip and palate phenotypes as part of a comprehensive search for aetiological mutations.
Several rare amino acid missense mutations are reported here as found in cleft cases. The qualitative evaluation of missense mutations using the Jalview program is a useful tool for classifying our findings. The qualitative comparison also benefits from the ability to inspect visually the local environment surrounding a given amino acid to judge tolerance of a change. By these measures, all five missense mutations found exclusively in cases are conserved phylogenetically to at least the cow, mouse, or rat protein sequence, while those found in cases and/or controls are not conserved past the human/cow comparison. Four of the five missense amino acid changes involve a change of amino acid hydrophobicity while the fifth, V114G, is found at a site of extreme evolutionary conservation. In addition, the hydrophilic substitutions found in cases appear to interrupt locally hydrophobic regions. The A23V and A34G changes that are found in cases and controls are changes within amino acid class and are changes from the locally redundant alanine.
Multiple lines of evidence suggest that MSX1 is a gene that promotes growth and inhibits differentiation.22,53,54 A disruption of growth by mutations in MSX1 could cause a lack of distal facial bud outgrowth and consequent primary or secondary palatal clefting, consistent with mouse models of the disease.8,55 Another indication that the missense mutations found in cases may be aetiological is the finding54 that an MSX1 protein lacking the N-terminal domain was not able to upregulate cyclin D1 and inhibit differentiation. Early differentiation in the progress zone of the facial processes could reduce outgrowth. All of our missense mutations were found on the N-terminal side of the homeodomain protein. Identification of these mutations also opens the door to additional studies of protein-protein interaction using proteins developed from each of the identified mutations.
The presence of an antisense transcript at the MSX1 locus22 reinforced the need to search non-coding portions of the gene. This antisense region was included in the sequencing reported here, including a 389 nucleotide region located in the single intron of MSX1, which has strong homology between humans and mice (80% at the nucleotide level). While some small regions of the 3‘UTR and 5‘UTR were sequenced in fewer than 100 subjects, four mutations were found that are probably aetiological in these non-coding areas. Future studies can investigate the role of these non-coding mutations on sense or antisense RNA stability, RNA protein interactions, or promoter activity.
The linkage disequilibrium previously reported suggests that there are more mutations to be found in MSX1, and so additional conserved regulatory regions will be important to identify in the future. Alternatively, one of the described polymorphic variants showing linkage disequilibrium, either by themselves (the intronic CA repeat) or in disequilibrium with that variant, might itself be the predisposing allele. Intronic microsatellites have been shown to influence gene expression suggesting functional analysis for this region will be important.
In this report, we showed that mutations can be identified in some subjects with non-syndromic cleft lip and palate. Supportive data, such as amino acid sequence conservation or mouse-human sequence homology, supports an aetiological role. Although it will remain for future studies to confirm that these particular mutations are causal, in the aggregate they would appear to contribute about 2% of all cases of non-syndromic cleft lip and palate. Five of our cases had other family members with clefts with the mutation segregating in three who were available for study. Some specific cases are thus likely to occur with a greater frequency where there is a family history of clefting, and so future clinical studies can be directed at identifying aetiologies by focusing particularly on these strongly positive families. Since the recurrence risks for sibs following the primary occurrence of cleft in the family range from 3-5%, identifying even 2% of cases that might have a Mendelian form in which recurrence could be as high as 50% has the potential for changing both recurrence risks for the population as a whole and certainly recurrence risks within these individual families. Although it may, as yet, be premature to include diagnostic MSX1 sequencing as part of the routine evaluation of non-syndromic cases of cleft lip and palate, this will be a consideration once future studies can further expand this finding.
Acknowledgments
The first two authors contributed equally to this work. We would particularly like to acknowledge the willing contribution of the many affected subjects and their family members who participated in this study. A large number of laboratory personnel and students have greatly assisted in this analysis including John Allaman, Kristin Aquilino, Ann Basart, Lisa Fascher, Jill Harrington, Naomi Lohr, Amy Luken, Marion Penning, and Min Shi. Lora Muilenburg assisted with administrative support. Control samples were contributed by Renata Lucia Ferreira de Lima. Kevin Knudtson was especially helpful. Operation Smile, Bill and Kathy Magee, and Edith Villanueva have been especially generous with their support. This work was supported in part by grants from the NIH, including P60 DE13076-02, DEO8559, ES10876, DE11948, and DK25295; D43 TW05503 from The Fogarty International Center; and U50/CCU 713238 from the CDC.
REFERENCES
Supplementary materials
. Web-only Appendices
Available as PDF (printer-friendly files)Files in this Data Supplement:
- [View PDF] - Total number of samples used in the study by population, cleft status, and gene region
- [View PDF] - Case-Control Frequency of the MSX1 Mutations and Polymorphisms Diagnosed in this Study
- [View PDF]