Article Text

Abstract
The spondylocostal dysostoses (SCD) are a group of disorders characterised by multiple vertebral segmentation defects and rib anomalies. SCD can either be sporadic or familial, and can be inherited in either autosomal dominant or recessive modes. We have previously shown that recessive forms of SCD can be caused by mutations in the delta-like 3 gene, DLL3. Here, we have sequenced DLL3 in a series of SCD cases and identified 12 mutations in a further 10 families. These include 10 novel mutations in exons 4–8, comprising nonsense, missense, frameshift, splicing, and in frame insertion mutations that are predicted to result in either the truncation of the mature protein in the extracellular domain, or affect highly conserved amino acid residues in the epidermal growth factor-like repeats of the protein. The affected cases represent diverse ethnic backgrounds and six come from traditionally consanguineous communities. In all affected subjects, the radiological phenotype is abnormal segmentation throughout the entire vertebral column with smooth outlines to the vertebral bodies in childhood, for which we suggest the term “pebble beach sign”. This is a very consistent phenotype-genotype correlation and we suggest the designation SCD type 1 for the AR form caused by mutations in the DLL3 gene.
- DLL3
- Notch signalling pathway
- somitogenesis
- spondylocostal dysostosis
Statistics from Altmetric.com
The spondylocostal dysostoses (SCD) are a heterogeneous group of disorders with severe axial skeletal malformation, characterised radiologically by multiple vertebral segmentation defects and rib anomalies, which are frequently malaligned with points of fusion and sometimes a reduction in number. These diverse SCD phenotypes can either be sporadic or familial.1 Monogenic SCD families have been reported with both autosomal dominant (AD)2–5 and autosomal recessive (AR) inheritance.6–13 Associated features in familial SCD include anal and urogenital anomalies,14 congenital heart disease,15,16 limb abnormalities,17 plagiocephaly-torticollis sequence,12 and inguinal herniae in males.12,18 Abnormal neurological signs and/or mental retardation are unusual. In sporadic cases clinical features are very heterogeneous and malformations beyond the vertebrae and ribs are more common.1,19,20 Nomenclature in this field is also confused by the inconsistent use of (varied) terminology. “Spondylo-thoracic” dysostosis (STD) has been used for the severe and often lethal AR form with a crab-like chest on radiograph, recently mapped to 2q32.1,21 but also sometimes for the mild AR form (similar to the mutation positive families of this study). “Spondylo-costal” dysostosis has been reserved by some for AD families,1 where the phenotype is generally mild, but also for the mild AR form.22 The term “costovertebral dysplasia” features in earlier reports.6,7,23,24 Many clinicians continue to use the eponymous Jarcho-Levin syndrome (JLS) across the spectrum of radiological phenotypes which include abnormal vertebral segmentation (AVS) and rib malalignment. The two sibs described by Jarcho and Levin25 had segmentation defects of the entire vertebral column, though most severe in the thoracic region, with fusion of several ribs. Both died in infancy of respiratory failure and the first born may also have had dextrocardia. With fusion of many vertebral bodies there were overlapping features with Klippel-Feil syndrome.
We have previously shown that some AR SCD families link to 19q13, a region syntenic with the locus of the mouse Dll3 gene, mutations in which result in the “pudgy” phenotype.26–28 Subsequent mutation analysis has determined that mutations in the human somitogenesis gene, delta-like 3, which encodes a ligand for the Notch signalling pathway, cause AR SCD.29 Human delta-like 3 is encoded by an eight exon gene that spans approximately 9.2 kb of chromosome 19. A 1.9 kb transcript encodes a protein of 618 amino acids. The protein consists of a signal sequence, a delta-serrate-lag2 (DSL, receptor interacting) domain, six epidermal growth factor (EGF)-like domains, and a transmembrane domain (TM).
In an attempt to clarify phenotype-genotype relationships we have sequenced DLL3 in a large cohort of subjects affected with AVS (both familial and sporadic), most cases also having rib anomalies. We describe our molecular findings and consistent clinical features in mutation positive cases, and suggest a rationale for the terminology.
SUBJECTS AND METHODS
Subjects
Cases were recruited through clinicians seeking DLL3 mutation analysis in their patients (see Acknowledgements). For the purposes of this comprehensive clinical-molecular presentation of DLL3 mutations, our tabulated data includes the three families (one Arab-Israeli, two originally Pakistani but resident in the UK) previously reported as part of our discovery of DLL3,12,29,30 as well as a novel mutation identified in a Palestinian Arab kindred.31 A further three UK Pakistani families were recruited, two of Turkish ethnicity, one Lebanese, and four families with mainly mixed European ancestry, including Ashkenazi Jewish in one case, though widely distributed geographically. Ethnic identity is incorporated in the tabulated mutation data (table 1).
Patient mutations detected in this study
Identification of mutations in the DLL3 gene
The DLL3 gene was amplified as previously described.29 Exon numbering for DLL3 follows that of the alternately spliced mouse Dll3 homologue, so the published human transcript begins with exon 2. PCR products were amplified using 50 ng genomic DNA in a 25 μl reaction mixture containing 10 pmol forward and reverse primers, 5 nmol each of dTTP, dCTP, and dATP, 3.8 nmol dGTP, 1.25 nmol deaza GTP, 1.25 μl DMSO, 25 μmol betaine, 10 mmol/l Tris-HCl, 50 mmol/l KCl, 1.5 mmol/l MgCl2, and 0.5 U Amplitaq Gold Taq polymerase (Applied Biosystems). After an initial denaturation of 95°C for 12 minutes, 40 cycles were performed which consisted of 95°C for one minute, 55°C for one minute, and 72°C for two minutes, with a final extension step of 72°C for 10 minutes. DNA fragments were purified using QIAquick purification columns (Qiagen) and sequenced using Big Dye Terminator Cycle sequencing (Applied Biosystems) according to the manufacturers’ recommendations. Sequencing products were separated by polyacrylamide gel electrophoresis on an ABI 377 DNA sequencer and analysed using FACTURA™ and Sequence Navigator 1.0.1. software (Applied Biosystems).
Haplotype analysis
Microsatellite markers proximal and distal to the Dll3 gene were amplified in a mix containing 40 ng genomic DNA, 5 pmol of forward and reverse primers, 2.5 nmol each dNTP, 10 mmol/l Tris-HCl, 50 mmol/l KCl, 1.5 mmol/l MgCl2, and 0.25 U Amplitaq Gold Taq polymerase (Applied Biosystems). Forward or reverse primers were labelled with 6-FAM, HEX, or TET fluorescent dyes (table 2). Reaction conditions were initial denaturation of 95°C for 12 minutes, followed by 10 cycles of 94°C for 30 seconds, 55°C for 30 seconds, 72°C for one minute, and then 30 cycles of 89°C for 30 seconds, 55°C for 30 seconds, and 72°C for one minute, with a final step of 72°C for seven minutes. PCR products were pooled in a 1:2:1 ratio of 6-FAM:HEX:TET labelled markers and mixed with GS-500 size standard (Applied Biosystems). Labelled products were separated by polyacrylamide gel electrophoresis on an ABI 377 DNA sequencer and analysed using Genescan 3.0 and Genotyper 2.5 software (Applied Biosystems).
Chromosome 19 markers used in haplotype nnalysis
Novel CA/GT repeat intergenic microsatellite markers (DLL3 CA1, GT1, GT2) were designed using the DNA sequence downloaded from the Human Genome Working Draft (http://genome.ucsc.edu/), and screened using the “Repeat Masker” software (http://www.hgmp.mrc.ac.uk/).
RESULTS
Clinical phenotype
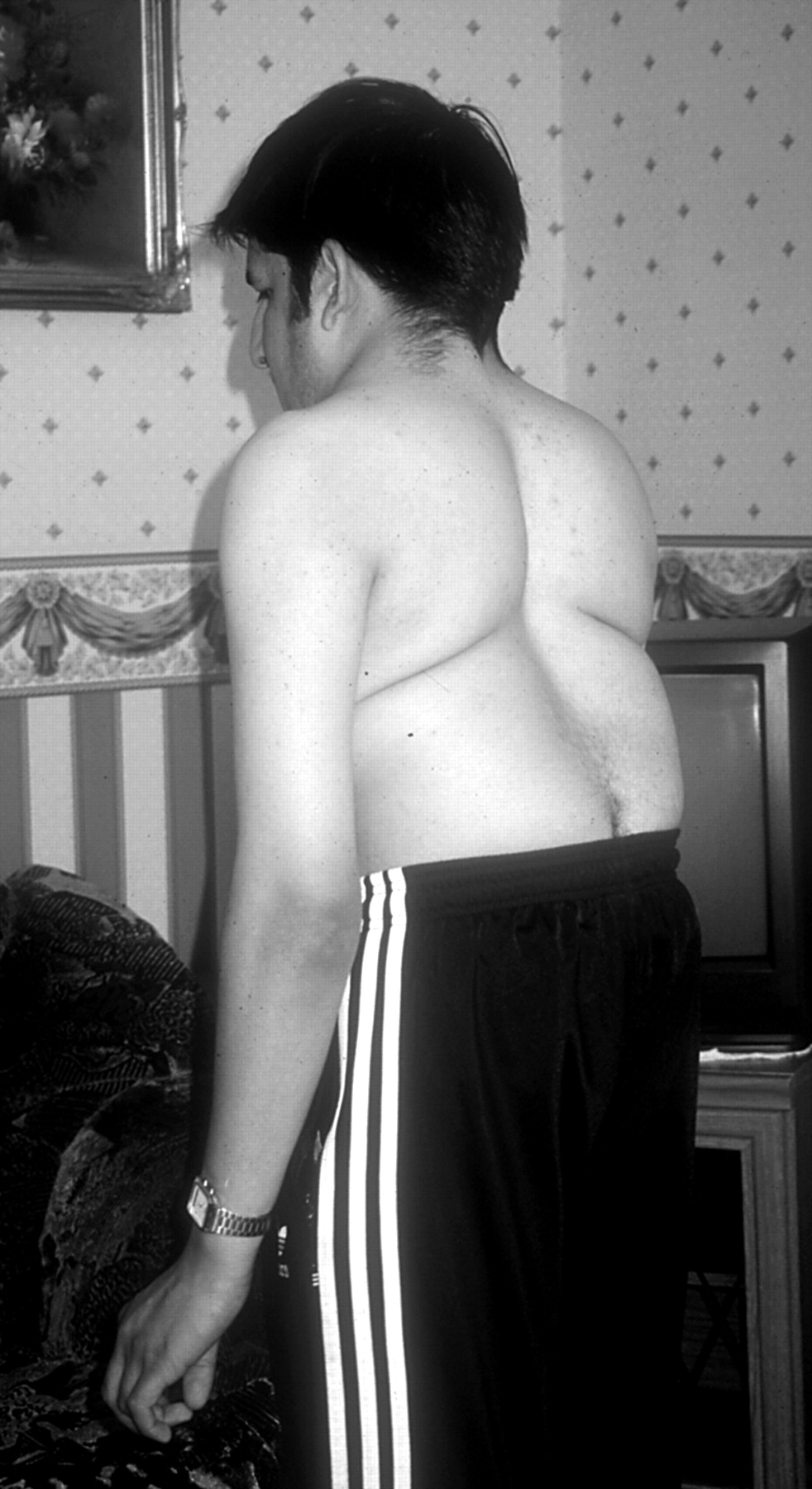
The majority of subjects, homozygous or compound heterozygous for mutations in DLL3, have truncal shortening relative to their limbs, which are otherwise normal. This gives rise to abdominal protrusion, abnormal spinal curvature (figs 11 and 2), and sometimes a plagiocephaly-torticollis sequence. In some reported families, inguinal herniae have been frequent in affected males,12 but it is not known whether this is a primary associated defect or a consequence of increased intra-abdominal pressure. The spinal curvature is a fixed, mild kyphoscoliosis that does not require corrective surgical fixation. Neurological complications in this form of SCD appear to be very rare. However, in this cohort, a notable exception is family 5 (table 1). In this heavily inbred family, two affected sibs had a form of congenital distal arthrogryposis and neurodevelopmental delay and are both now dead. It is not known whether these additional severe features were the result of mutated DLL3 or represent a separate autosomal recessive condition giving rise to fetal akinesia sequence.

The general appearance of an adult with SCD. The upper limbs have the appearance of being excessively long but this is the result of truncal shortening. There is abdominal protrusion.

The general appearance of an adult with SCD, posterior view of the same subject as illustrated in fig 1. There is truncal shortening with the appearance of an exaggerated mid-thoracic lordosis.
Spinal radiographs were reviewed for all affected cases. Typical examples are shown in figs 3–5. The radiological phenotype is consistent, comprising multiple hemivertebrae throughout the entire spinal column. The nature of the abnormal segmentation is similar in all regions from cervical to lumbar, and sacral morphology is more difficult to discern, but present. In childhood, before full maturity of the spine, the shape of the abnormal vertebrae is ovoid or circular in the majority and the contours smooth. In all cases the ribs are irregularly aligned and points of fusion occur to a variable degree.

The radiological appearances of SCD resulting from mutated DLL3 in a fetus terminated at 20 weeks’ gestation. Note the irregular alignment of the vertebral bodies throughout the spine as well as abnormally aligned ribs with points of fusion.

The radiological appearances of SCD resulting from mutated DLL3 in an infant. Note the irregular alignment of the vertebral bodies throughout the spine and the smooth contours to the vertebrae. The ribs are abnormally aligned with asymmetrical points of fusion.

The radiological appearances of SCD resulting from mutated DLL3 in a child of 1 year. Mild scoliosis has developed (compared with fig 4) but this will not progress significantly. The vertebral bodies show the same characteristic smooth contours as seen in fig 4, for which we propose the radiological term “pebble beach sign”.
Molecular genetic analysis
Direct sequencing of the DLL3 gene from genomic DNA showed mutations in a further 10 AR SCD families. The data presented here brings to 17 the number of families now reported.32 A total of 17 different mutations have now been found, 10 of which are described here for the first time. The novel mutations include six frameshift (395delG, 602delG, 614ins13, 948delTG, 1365del17, 1418delC), two nonsense (C207X, C362X), one splicing (868del11), and one in frame insertion (1256ins18). The mutation 395delG in exon 4 occurs within the amino terminal domain of the protein and is predicted to result in a truncated protein lacking the delta-serrate-lag2 domain, all six EGF-like repeats and the transmembrane and intracellular domains. The mutations 602delG, 603ins5, 614ins13, and 621C>A in exon 5 occur within the delta-serrate-lag2 domain and may result in proteins lacking all the six EGF repeats and the transmembrane and intracellular domains. The mutations 945delAT and 948delTG in exon 7 occur within EGF repeat 3 and would result in the loss of EGF repeats 4–6 and the transmembrane and intracellular domains. The mutation C362X in exon 7 occurs within EGF repeat 4 and would result in the loss of EGF repeats 5–6 and the transmembrane domain. The mutations 1365del17 and 1418delC occur within or after EGF repeat 6, respectively, and would both lead to the loss of the transmembrane and intracellular domains. The previously reported missense mutation C309Y in exon 731 occurs within EGF repeat 2 and results in the substitution of one of the six cysteine residues required in each EGF repeat and replaces it with an aromatic tyrosine residue. This would prevent the correct folding of EGF repeat 2 and disable normal function. The C309Y mutation was not found in 31 ethnically matched controls (62 chromosomes). The 868del11 mutation removes the donor splice site of intron 6 and is likely to cause aberrant splicing. The in frame insertion 1256ins18 in exon 8 inserts the six amino acid peptide His-Arg-Cys-Ser-Cys-Ala into EGF repeat 5. This increases the number of cysteine residues in this repeat from 6 to 8 and would result in the incorrect folding of the EGF repeat domain. The mutations are characterised schematically in the DLL3 gene in fig 6. In addition, a range of polymorphisms have been characterised in the coding region and these are presented in table 3.
Polymorphisms identified in human DLL3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the human DLL3 mutations. The protein is encoded by eight exons of the DLL3 gene which spans ¬9.2 kb; the transcript encodes 618 amino acids. The protein consists of a delta-serrate-lag2 region (DSL), six epidermal growth factor-like domains (EGF), and a transmembrane domain (TM).
Haplotype analysis
The exon 5 mutation 603ins5 has been observed in SCD families 1 and 7, but are not known to be related. Haplotype and intragenic polymorphism analysis showed two different haplotypes (data not shown) suggesting that the mutation 603ins5 is not the result of a single mutation in a common ancestor. The occurrence of this mutation can be explained by template slippage during replication owing to the presence of tandem repeats of the sequence GCGGT. The mutation 614ins13 in exon 5 was seen in families 11 and 12. However, in this case haplotype analysis showed the high likelihood of a single common ancestral mutation (data not shown). The mutation 945delAT in exon 7 has been observed in SCD families 2 and 4, and again haplotype analysis showed the high likelihood of a single common ancestral mutation (data not shown).
DISCUSSION
Abnormal vertebral segmentation (AVS) is a significant developmental malformation in clinical dysmorphology and is a feature, to a very variable degree, of a large number of syndromes and associations (table 4). However, forms of AVS showing clear Mendelian inheritance are relatively rare and, as table 4 highlights, the cause of most of these syndromic conditions and associations is unknown. AVS is most frequently seen in sporadic cases with diverse radiological phenotypes, which are difficult both to classify1 and investigate. Some may be the result of chromosome abnormalities; AVS is a well recognised feature of trisomy 8 mosaicism33 and has also been reported with multiple congenital anomalies in association with other abnormal karyotypes, both (apparently) balanced34 and unbalanced.35–37 Clarke et al38 have described a four generation pedigree with pericentric inversion inv(8)(q22.2q23.3) cosegregating with cervical vertebral anomalies of Klippel-Feil syndrome.
Some syndromes and conditions which include abnormal vertebral segmentation
The Notch signalling pathway is required for normal somite formation during embryonic development. Vertebrae are derived from somites and therefore mutations in genes associated with the Notch signalling pathway are implicated in the genesis of AVS. Notch is a large transmembrane receptor that binds to two distinct families of ligand, delta and jagged/serrate.39 The affinity of Notch for these ligands can be modified by differential glysosylation mediated by the fringe protein, encoded by the glycosyl transferase, lunatic fringe.40,41 This “cycling gene” is expressed as a wave that sweeps rostrally across the presomitic mesoderm from the caudal end. This occurs in early development with precise periodicity with each cycle culminating in the segmentation of a somite from the presomitic mesoderm. Lunatic fringe is thus considered a molecular read out of a “segmentation clock”. The Notch signalling pathway is intrinsic to the oscillations of the segmentation clock. Disruption of Notch signalling leads to perturbation of the clock and aberrant somite formation. For example, mice that lack delta-like 3 display AVS and rib malformations as a result of delayed and irregular somite formation in the embryo. This is the result of disruption of the segmentation clock since expression of cyclical genes (lunatic fringe, hes1, and hes5) is abnormal.27 However, it is not yet clear exactly how Notch signalling leads to somite boundary formation.42 In addition to mutations in delta-like 3 causing SCD, mutations in jagged1 cause the autosomal dominant Alagille syndrome (arteriohepatic dysplasia), a multiple congenital abnormality condition with butterfly vertebrae in 64% of patients.43–45
This is the first substantial cohort of cases with AVS investigated by sequencing of the delta-like 3 gene, DLL3. The findings show that mutations in DLL3 are associated with a consistent radiological phenotype (figs 3–5). Abnormal segmentation occurs throughout the entire spinal column and the shape and contours of the abnormal vertebrae resemble smooth, eroded pebbles such as those found on shingle or stony beaches. We suggest this distinctive pattern could be identified in skeletal radiology by the term “pebble beach sign”. We have identified a total of 14 mutations in 14 families world wide, including four previously published.29,31 Eleven of these families are consanguineous, showing homozygosity for the DLL3 mutation in SCD subjects, while three are non-consanguineous and affected subjects are compound heterozygotes. Three further DLL3 mutations have been identified in SCD cases from a small community in Switzerland,32 bringing to 17 the total number of identified mutations to date.
Analysis of DLL3 mutation data (table 1) indicates that a significant proportion of cases are caused by familial mutations that are predicted to lead to premature termination of the delta-like 3 protein with the subsequent loss of important domains such as EGF repeats and/or the transmembrane domain. This shows that the normal function of delta-like 3 during somitogenesis requires that the protein be anchored into the membrane in the correct orientation. Although some protein may be translated from these truncated alleles, mutant mRNA transcripts may be subject to degradation via the nonsense mediated mRNA decay pathway (reviewed by Frischmeyer and Dietz46). The missense mutation C309Y in EGF repeat 2 and the insertion of the six amino acid repeat HRCSCA into EGF repeat 5, as well as G385D in EGF repeat 4 previously characterised,29 indicate that the epidermal growth factor like repeats are also essential for the correct function of delta-like 3 during somitogenesis. In humans, the protein truncating and missense DLL3 mutations have essentially the same phenotypic effect as the null Dll3 mutation generated in mouse. This suggests that each of the DLL3 mutations reported here are likely to be null or severely hypomorphic. Of interest, the normal phenotype of heterozygous subjects indicates that none of the mutations characterised to date acts in a dominant negative manner or through haploinsufficiency of the protein. One heterozygous subject in family 1 (described with images in Turnpenny et al12,30) has mild mid-thoracic scoliosis and AVS in the lower lumbar vertebrae. However, we cannot be certain at present whether this is causally linked to her DLL3 heterozygous status. In general, therefore, the mutations characterised here would all appear to be pure loss of function mutations and are only recessively inherited.
While the classification of the spondylocostal dysostoses remains difficult, we propose that the term spondylocostal dysostosis (SCD) be reserved for the radiological phenotype characterised by irregular formation of all vertebrae, usually in association with abnormally aligned ribs showing points of fusion. Cases showing AVS in restricted regions of the spine would therefore be excluded from this diagnostic group. We have shown that DLL3 is the most commonly mutated gene in SCD. We further propose that the distinctive “pebble beach” radiological sign, which appears always to result from mutated DLL3, be designated SCD “type 1”.
Acknowledgments
We are grateful to Action Research for financial support (project grant No SP3751 - PDT, SE, and NW), to the Royal Devon and Exeter National Healthcare Trust, the University of Exeter and the Peninsula Medical School, and the NH&MRC (project grant No 142006, SLD). We thank Michael Tracy for technical assistance. KK is a Hitchings-Elion Fellow of the Burroughs Wellcome Fund. The following clinicians made this study possible by generously enrolling their patients: Juan Bernar (Madrid, Spain), Lynne Bird, (San Diego, USA), Nursel Elcioglu (Istanbul, Turkey), Christine Garrett (Kennedy-Galton Centre, Middlesex, UK), Amanda Krause (Johannesburg, South Africa), Meriel McEntagart (Kennedy-Galton Centre, Middlesex, UK), Carole McKeown (Birmingham, UK), Steven Munjanja (Harare, Zimbabwe), David Sillence (Sydney, Australia), Joep Tuerlings, (Nijmegen, The Netherlands), and Geoff Woods (Leeds, UK).