Article Text

Abstract
Leigh syndrome is a subacute necrotising encephalomyopathy frequently ascribed to mitochondrial respiratory chain deficiency. This condition is genetically heterogeneous, as mutations in both mitochondrial (mt) and nuclear genes have been reported. Here, we report the G13513A transition in the ND5 mtDNA gene in three unrelated children with complex I deficiency and a peculiar MRI aspect distinct from typical Leigh syndrome. Brain MRI consistently showed a specific involvement of the substantia nigra and medulla oblongata sparing the basal ganglia. Variable degrees of heteroplasmy were found in all tissues tested and a high percentage of mutant mtDNA was observed in muscle. The asymptomatic mothers presented low levels of mutant mtDNA in blood leucocytes. This mutation, which affects an evolutionary conserved amino acid (D393N), has been previously reported in adult patients with MELAS or LHON/MELAS syndromes, emphasising the clinical heterogeneity of mitochondrial DNA mutations. Since the G13513A mutation was found in 21% of our patients with Leigh syndrome and complex I deficiency (3/14), it appears that this mutation represents a frequent cause of Leigh-like syndrome, which should be systematically tested for molecular diagnosis in affected children and for genetic counselling in their maternal relatives.
- G13513A
- Leigh disease
- NADH
- mitochondrial DNA
Statistics from Altmetric.com
Leigh syndrome is a neuropathological entity characterised by symmetrical necrotic lesions along the brainstem, diencephalon, and basal ganglion. This condition is frequently accounted for by respiratory chain deficiency in early childhood. Deficiencies of complex I (NADH ubiquinone reductase), complex II (succinate ubiquinone reductase), complex IV (cytochrome oxidase), and complex V (ATPase) of the mitochondrial respiratory chain have been reported in Leigh syndrome.1 This condition is genetically heterogeneous as both mitochondrial and nuclear mutations have been reported: mutations in nuclearly encoded subunits of complex I (NUDFV1 and NDUFS1),2,3 complex II (SDH Fp),4 mutations in assembly proteins of complex IV (SURF1),5,6 and a mutation in a mitochondrial (mt) subunit of complex V (NARP mutation in ATPase 6).7 Recently, two heteroplasmic mutations involving mitochondrially encoded complex I subunits have been identified in patients with Leigh syndrome and complex I deficiency, namely a Leber (LHON)/dystonia mutation in the ND6 gene (G14459A)8 and a novel missense mutation in the ND5 gene (T12706C).9 Here, we show that the G13513A mutation in the mitochondrial ND5 gene previously reported in adult MELAS or LHON/MELAS patients was found in 3/14 infants with Leigh-like syndrome and complex I deficiency in our series. We suggest that this mutation should be regarded as a frequent cause of Leigh-like syndrome in infancy.
PATIENTS AND METHODS
Patient 1
A boy was born to unrelated, healthy parents after a 34 week pregnancy. He was small for gestational age (weight 2000 g, length 44 cm, OFC 32 cm). Two older sibs are healthy but one sister had severe intrauterine growth retardation and died suddenly at 22 hours of age. He had two attacks of regression, drowsiness, truncal hypotonia, and metabolic acidosis at 6 and 11 months of age respectively. Developmental milestones were markedly delayed and severe failure to thrive required nasal tube feeding. Brisk deep tendon reflexes, squint, paralysis of vertical gaze, and transient hypertrophic cardiomyopathy were noted at 2 years of age. His plasma (3.5 mmol/l) and CSF lactate were raised (6.5 mmol/l, control below 2.4 mmol/l). Brain MRI showed T1 hypointensity of the substantia nigra (fig 1A).
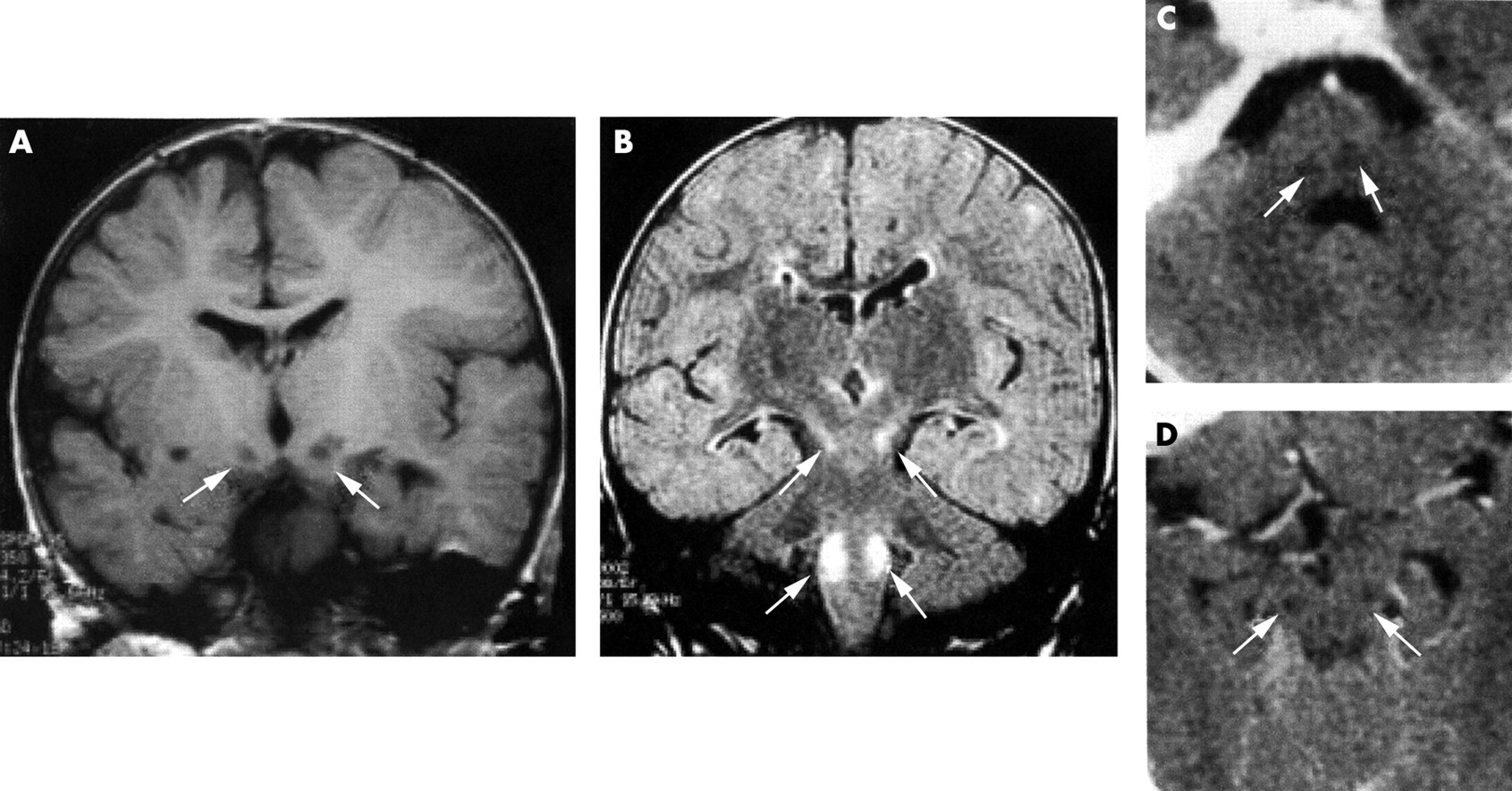
MRI of patients. (A) Patient 1. T1 weighted coronal image showing bilateral hyposignal of the substantia nigra. (B) Patient 2. FLAIR coronal image showing bilateral hypersignal of the substantia nigra and the dorsal columns of the medulla oblongata. (C, D) Patient 3. Axial CT scan images at the level of the medulla oblongata (C) and the midbrain (D) showing bilateral hypodensities of the dorsal columns of the medulla oblongata and the substantia nigra.
Patient 2
A boy was born to unrelated, healthy parents after a 38 week pregnancy (weight 2700 g, length 46 cm, OFC 32 cm). Mild dysmorphic features included coarse facies, bilateral squint, unilateral cleft lip (operated on at 6 weeks of age), short distal phalanges, rostral vertebrae (L1), and a single palmar crease. Severe failure to thrive and psychomotor retardation remained unexplained. At 21 months of age, he suddenly developed an attack of regression, drowsiness, vomiting, myoclonic jerks, massive hypotonia, hypothermia, bradypnoea, and bradycardia suggestive of severe brainstem involvement. A severe hypertrophic cardiomyopathy was first observed at that time but no other organ involvement was noted (CSF lactate 3.8 mmol/l, normal below 2.4 mmol/l). Brain MRI showed bilateral T2 hyperintensities in the substantia nigra and the dorsal column of the medulla oblongata (fig 1B).
Patient 3
A boy was born to unrelated, healthy parents after a 35 week pregnancy. Severe intrauterine growth retardation was ascribed to toxaemia gravidis (weight 1680 g, length 43 cm, OFC 30 cm). His maternal aunt had died at 4 months of age of an unexplained bout of pallor, cyanosis, and massive hypotonia. His older sister is healthy. Mild dysmorphic features included round face, frontal bossing, flat nasal root, long, featureless philtrum, bilateral ptosis, squint, progressive external ophthalmoplegia, thin lips, small chin, hypospadias, and inguinal hernia. Failure to thrive (weight −4 SD, height −3 SD), swallowing difficulties, massive hypotonia, and muscular atrophy required nasal tube feeding at 24 months of age. He gained weight significantly and his psychomotor development improved but he could not sit unaided or follow with his eyes. Retinal dystrophy with extinguished electroretinogram was noted but no other organ involvement was observed. At 21 months, he suddenly developed an attack of regression, drowsiness, vomiting, diarrhoea, myoclonic jerks of the limbs and eyelids, bradycardia, and bradypnoea. Metabolic acidosis (bicarbonate 15 mmol/l), raised plasma lactate (3.5-6.0 mmol/l, normal below 2.4 mmol/l), and urinary excretion of Krebs cycle intermediates were consistently noted. CT scan disclosed bilateral hypodensities of the substantia nigra and dorsal medulla oblongata (fig 1C, D).
Polarographic and spectrophotometric studies on muscle mitochondria were performed as previously described.10 Skin fibroblasts were grown in a RPMI medium supplemented with glutamax (446 mg/l), 10% undialysed fetal calf serum, 100 μg/ml streptomycin, 100 IU/ml penicillin, glucose (2 g/l), 200 μmol/l uridine, and 2.5 mmol/l sodium pyruvate.11 Genomic DNA from skeletal muscle, cultured skin fibroblasts, and/or blood leucocytes was prepared according to standard procedures.
RESULTS
Activity ratios involving complex I were significantly abnormal in patient 1 (table 1). Similarly, polarographic and spectrophotometric studies on muscle mitochondria of patients 2 and 3 showed decreased complex I activities and abnormal activity ratios, confirming the diagnosis of complex I deficiency (table 1).
Enzymological investigations in skeletal muscle and cultured skin fibroblasts of Leigh disease patients
Neither large scale mtDNA rearrangements, nor mutations in the nuclear genes of complex I (NDUFV1, NDUFS8, NDUFS7, NDUFB6, NDUFA8, NDUFS1, NDUFS4, NDUFS2, NDUFV2) were found in the three patients. PCR amplification and sequence of the mitochondrial ND5 gene12 in skeletal muscle of the three patients detected a G to A transition at nucleotide 13513, changing an aspartic acid into an asparagine in a highly conserved region of the protein (D393N).
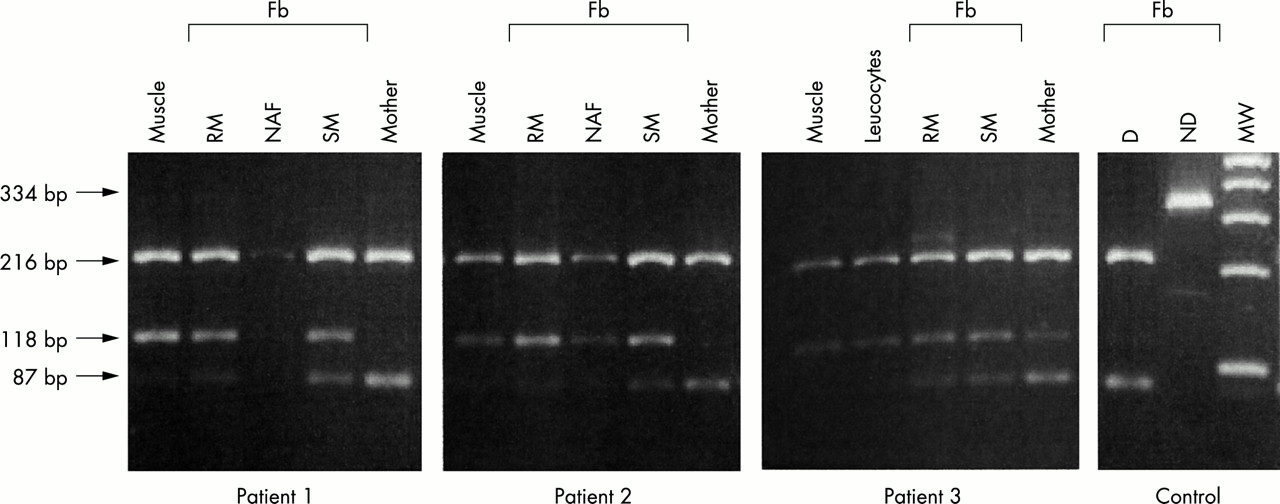
PCR amplification using the allele created restriction site technique showed a high level of heteroplasmy in several tissues. Indeed, heteroplasmy for the G13513A mutation averaged 90% in skeletal muscle of the three patients. The level of heteroplasmy was very high in circulating lymphocytes of patient 3, a result which is consistent with weak oxidation of pyruvate in his lymphocytes (15 nmol/min/mg protein, control 24-94 nmol/min/mg protein). Low levels of the G13513A mutation were found in lymphocytes of the asymptomatic mothers (5-20%, fig 2).
{kind=link}
{kind=link}
Quantification of the G13513A ND5 gene mutation. To evaluate the level of heteroplasmy, PCR amplification of the ND5 gene was performed using a mismatched forward primer CCTCACAGGTTTCTACTCCGAA (nt 13491-nt 13512) and a reverse primer (nt 13825-nt 13806), which generated a 334 bp fragment. Digestion of wild type mtDNA with restriction enzyme MboII generated three fragments of 216, 87, and 31 bp, while digestion of the mutant mtDNA generated only two fragments of 216 and 118 bp in length. Fb: fibroblasts grown either in regular medium (RM) or in selective medium (SM). NAF: non-adhering fibroblasts in selective medium. D: digested control DNA. ND: non-digested control DNA. MW: molecular weight.
Large amounts of mutant mtDNA molecules (80-90%) were also observed in skin fibroblasts of patients 1 and 2. Interestingly, the level of heteroplasmy for the G13513A mutation was much lower when the patient’s fibroblasts were grown in a selective medium devoid of glucose, uridine, and pyruvate (fig 2). Similarly, the patient’s fibroblasts expressed complex I deficiency when grown in standard conditions while this activity returned to normal levels when the fibroblasts were grown in a selective medium (table 1). This observation should be ascribed to the counter-selection of cells harbouring high levels of mutant mtDNA molecules in the selective medium. In fact, a number of floating cells carrying 80-95% of mutant mtDNA molecules were observed when fibroblasts were grown in this selective medium (fig 2). The level of heteroplasmy was lower in patient 3 (60%), regardless of the culture conditions.
DISCUSSION
Leigh syndrome is a well characterised neuropathological entity characterised by the distribution of symmetrical necrotic lesions along the brainstem, diencephalon, and basal ganglion. Interestingly, neuroimaging of the three children (MRI or CT scan) showed a selective brainstem involvement of the substantia nigra and dorsal columns of the medulla oblongata sparing the basal ganglia. Owing to this atypical neuroradiological pattern, we refer to Leigh-like syndrome in our patients.
The detection of the G13513A mutation in the mitochondrial ND5 gene of infants and children with Leigh-like syndrome and complex I deficiency is of particular importance for several reasons. First, the detection of this mutation in 3/14 unrelated cases of Leigh-like syndrome and complex I deficiency and in none of 35 additional complex I deficient patients with other clinical presentations (not shown) strongly suggests that this mutation is a major cause of Leigh/Leigh-like syndrome (21% of cases of Leigh/Leigh-like syndrome and complex I deficiency in our series). We suggest that children with Leigh syndrome and complex I deficiency should be systematically screened for the G13513A mtDNA mutation and the other two mutations involving mtDNA encoded complex I subunits8,9 before further screening of nuclear encoded complex I subunit genes. Given that the disease was maternally inherited from asymptomatic carrier mothers, screening for the G13513A mtDNA mutation has a major impact on the genetic counselling of maternal relatives.
A second aspect of this study is the potential use of culture medium for determining the deleterious nature of a mtDNA mutation. Indeed, the G13513A was heteroplasmic in all tested tissues, a high level of mutant mtDNA being observed in tissues expressing complex I deficiency. Growing complex I deficient fibroblasts in a selective medium (devoid of glucose, uridine, and sodium pyruvate) resulted in the counter-selection of defective cells, the remaining cells harbouring lower amounts of mutant mtDNA with normalised complex I activity. By comparing the ratio of mutant to normal mtDNA in fibroblasts grown in standard and selective media, we were able to determine the threshold of expression of complex I deficiency in these cells (70-80%).
Interestingly, intrauterine growth retardation and mild dysmorphic features, either isolated (patient 1) or associated with prenatal anomalies of development, were observed in the three children (patients 2 and 3). While most respiratory chain deficient children have normal birth weight and prenatal development, a fraction of them show a combination of intrauterine growth retardation and various malformations.13 These features should be related to the time of respiratory enzyme expression in the embryonic fetal period. The earlier the prenatal expression of the G13513A mutation, the earlier the developmental manifestations of the disease. In this case, our observation supports the early prenatal expression of the G13513A mutation in the three children.
The G13513A mutation has been previously described in patients with entirely different clinical presentations, namely adult onset MELAS,14 LHON/MELAS syndromes,15 and adult onset encephalomyopathy with blindness.16 Our results and a personal communication9 show that this mutation also accounts for Leigh disease in early infancy. Thus, the present study not only emphasises the striking clinical variability of mtDNA mutations, but also adds functional evidence for the deleterious nature of the G13513A mutation. Future reports will help to decide whether or not the involvement of the substantia nigra and medulla oblongata is specific to the G13513A ND5 gene mutation.
In conclusion, the G13513A mutation in the ND5 gene is an important and novel cause of Leigh-like syndrome and complex I deficiency in childhood. The systematic screening of all mitochondrially encoded complex I subunits will help to elucidate the actual impact of mtDNA mutations in Leigh syndrome with complex I deficiency.