Article Text

Abstract
Background DYNC1H1 encodes the heavy chain protein of the cytoplasmic dynein 1 motor protein complex that plays a key role in retrograde axonal transport in neurons. Furthermore, it interacts with the LIS1 gene of which haploinsufficiency causes a severe neuronal migration disorder in humans, known as classical lissencephaly or Miller-Dieker syndrome.
Aim To describe the clinical spectrum and molecular characteristics of DYNC1H1 mutations.
Methods A family based exome sequencing approach was used to identify de novo mutations in patients with severe intellectual disability.
Results In this report the identification of two de novo missense mutations in DYNC1H1 (p.Glu1518Lys and p.His3822Pro) in two patients with severe intellectual disability and variable neuronal migration defects is described.
Conclusion Since an autosomal dominant mutation in DYNC1H1 was previously identified in a family with the axonal (type 2) form of Charcot- Marie-Tooth (CMT2) disease and mutations in Dync1h1 in mice also cause impaired neuronal migration in addition to neuropathy, these data together suggest that mutations in DYNC1H1 can lead to a broad phenotypic spectrum and confirm the importance of DYNC1H1 in both central and peripheral neuronal functions.
- DYNC1H1
- intellectual disability
- neuronal migration disorder
- peripheral neuropathy
- genetics
- microRNA
- neurosciences
- chromosomal
- copy-number
- molecular genetics
- microarray
- metabolic disorders
- rheumatoid arthritis
- rheumatology
- renal medicine
- calcium and bone
- diagnostics tests
- genetic screening/counselling
- visual development
- clinical genetics
- academic medicine
- neuromuscular disease
- memory disorders
- hydrocephalus
- cytogenetics
Statistics from Altmetric.com
- DYNC1H1
- intellectual disability
- neuronal migration disorder
- peripheral neuropathy
- genetics
- microRNA
- neurosciences
- chromosomal
- copy-number
- molecular genetics
- microarray
- metabolic disorders
- rheumatoid arthritis
- rheumatology
- renal medicine
- calcium and bone
- diagnostics tests
- genetic screening/counselling
- visual development
- clinical genetics
- academic medicine
- neuromuscular disease
- memory disorders
- hydrocephalus
- cytogenetics
Neuronal migration disorders are an heterogeneous group of neurodevelopmental disorders that represent an important cause of intellectual disability (ID) and epilepsy in humans. Lissencephaly is due to failure of migration of neuronal precursors from the paraventricular zone to the cerebral cortex during early brain development.1–3 Lissencephaly is classified in several subtypes, one of which is classical lissencephaly.1 Classical lissencephaly can be present as an endophenotype in contiguous gene syndromes, such as Miller Dieker syndrome (deletion chromosome 17p13) (MDLS (MIM 247200)), but can also be present in an isolated form. The vast majority of classical lissencephaly is caused by haploinsufficiency of the Lissencephaly 1(LIS1)/platelet-activating factor acetylhydrolase, isoform 1B, α subunit (PAFAH1B) (MIM 601545) gene or mutations in the gene doublecortin (DCX (MIM 300121)) on the X-chromosome.1 4 Patients with classical lissencephaly have severe ID and develop intractable epilepsy. In addition they may present with early hypotonia which evolves in spasticity.1–3
The LIS1 protein plays a key role in cell migration of neuronal progenitors during brain development. LIS1 interacts with platelet activating factor acetylhydrolase and cytoplasmic dynein. By binding to cytoplasmic dynein LIS1 regulates microtubule dependent cell motility. This process is disrupted in lissencephaly.2 3 5 Neuronal cell migration is processed by nucleokinesis, which involves an extension of the neuron into which the nucleus is translocated via microtubule transport. It was shown that the cytoplasmic dynein 1 motor protein complex is the driving motor force of this process. Thus, LIS1 stimulates cytoplasmic dynein functions that involve neuronal migration and axon growth.2 3 5
Recently, we reported a de novo missense mutation in dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1 (MIM 600112)), a cytoplasmic dynein 1 motor complex gene, in a patient with ID as part of a larger study using a family based exome sequencing approach.6 Here we report the identification of a second de novo mutation in this gene in another patient with a distinct phenotype which overlaps with the phenotype of the patient in the previous study. These data together suggest that de novo mutations in DYNC1H1 cause variable phenotypes including severe ID with variable neuronal migration defects, and peripheral neuropathy.
Patient 1 was previously reported by Vissers et al.6 In brief, this boy was the first child born at term after an uneventful pregnancy with a weight of 3850 g (60th centile) to healthy parents. At 6 months hypotonia was noted and physical therapy was started. Development was delayed. He could sit at 18 months and walk at 3 years. At the age of 4 years his developmental level was tested which showed a 2 year delay, consistent with moderately severe ID. At the age of 6 years 6 months he could only speak several single words. On physical examination at the age of 2 years he had a normal height of 85.2 cm (15th centile) and head circumference of 47.5 cm (15th centile). He had mild (facial) dysmorphism consisting of prominent forehead, plagiocephalic skull, a hypotonic face with downslanted palpebral fissures, and short broad hands and feet. On further physical examination at the age of 6 years 6 months he had a normal height (122 cm (25–50th centile)) and head circumference (53.5 cm (84th centile)), and similar dysmorphism as on previous examinations.
Abnormal neurologic findings included hypotonia, general reduced tendon reflexes, and broad-based waddling gait with toe walking. Re-evaluation of the brain MRI that was performed at the age of 5 years 6 months showed, despite its suboptimal quality, signs of bilateral cortical malformation with deficient gyration of the frontal lobes (without clear abnormalities of cortical thickness) and an area suggestive of focal cortical dysplasia (figure 1A–C). A family based exome sequencing approach revealed a de novo c.11465A→C mutation in DYNC1H1 (NM_001376.4), predicting a p.(His3822Pro) substitution (figure 2A).6 Although the Grantham score at 77 indicates a moderate physicochemical difference, the PhyloP score at 5.12 suggests a very high conservation of the affected nucleotide among 46 vertebrate species. A prediction of the effect of this amino acid substitution at protein level by SIFT (sorting intolerant from tolerant) indicates that this mutation is deleterious to normal protein function.7
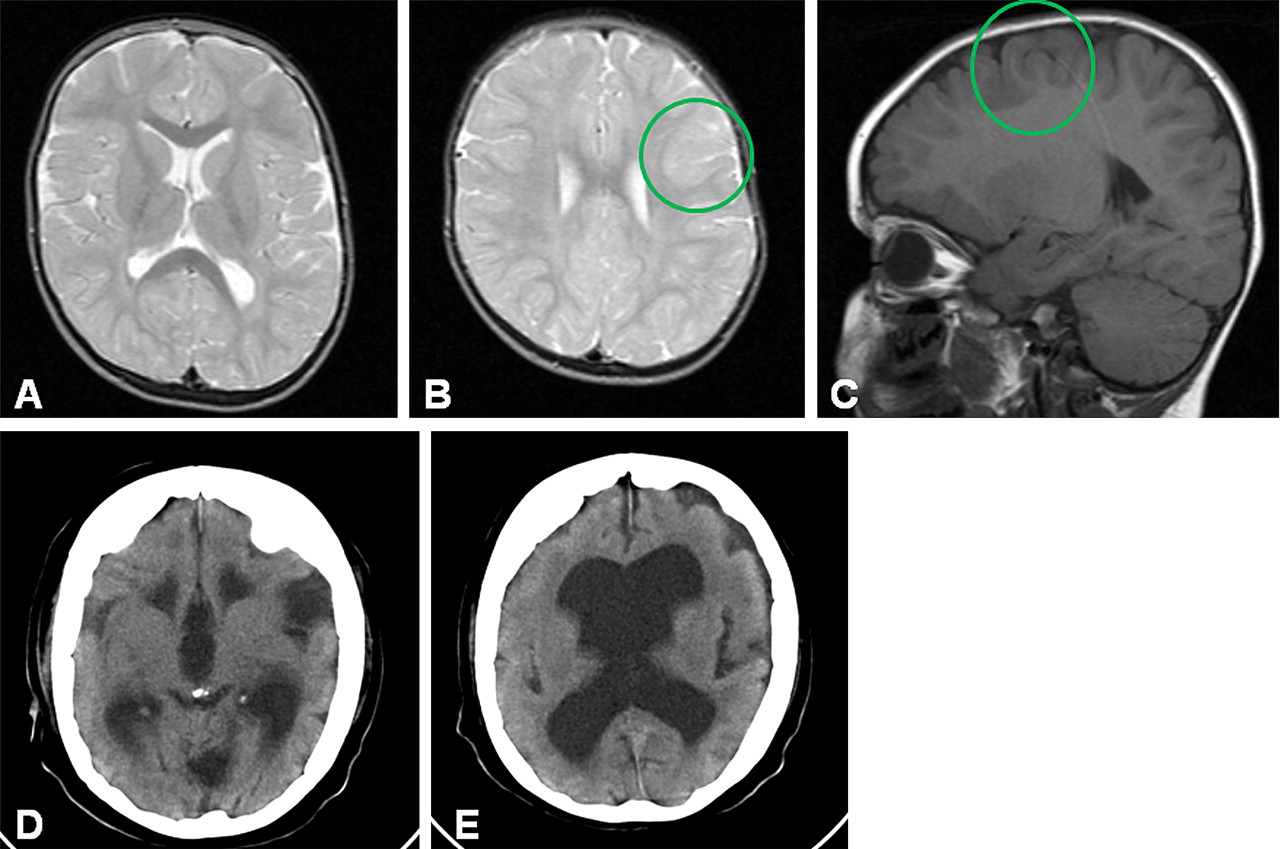
Cerebral imaging in patient 1 (A–C) and patient 2 (D, E). Panels A–C show axial, T2 weighted (A, B) and sagittal, T1 weighted (C) MR images with deficient gyration of the frontal lobes and an area suspected of being cortical dysplasia in patient 1 (circles in panels B and C). Panels D and E show CT images from patient 2, with enlarged ventricles, wide opercular regions, and clear signs of frontal lobe cortical malformation with an abnormal flat cortex with only a few, simple and shallow sulci.

{kind=link}
{kind=link}
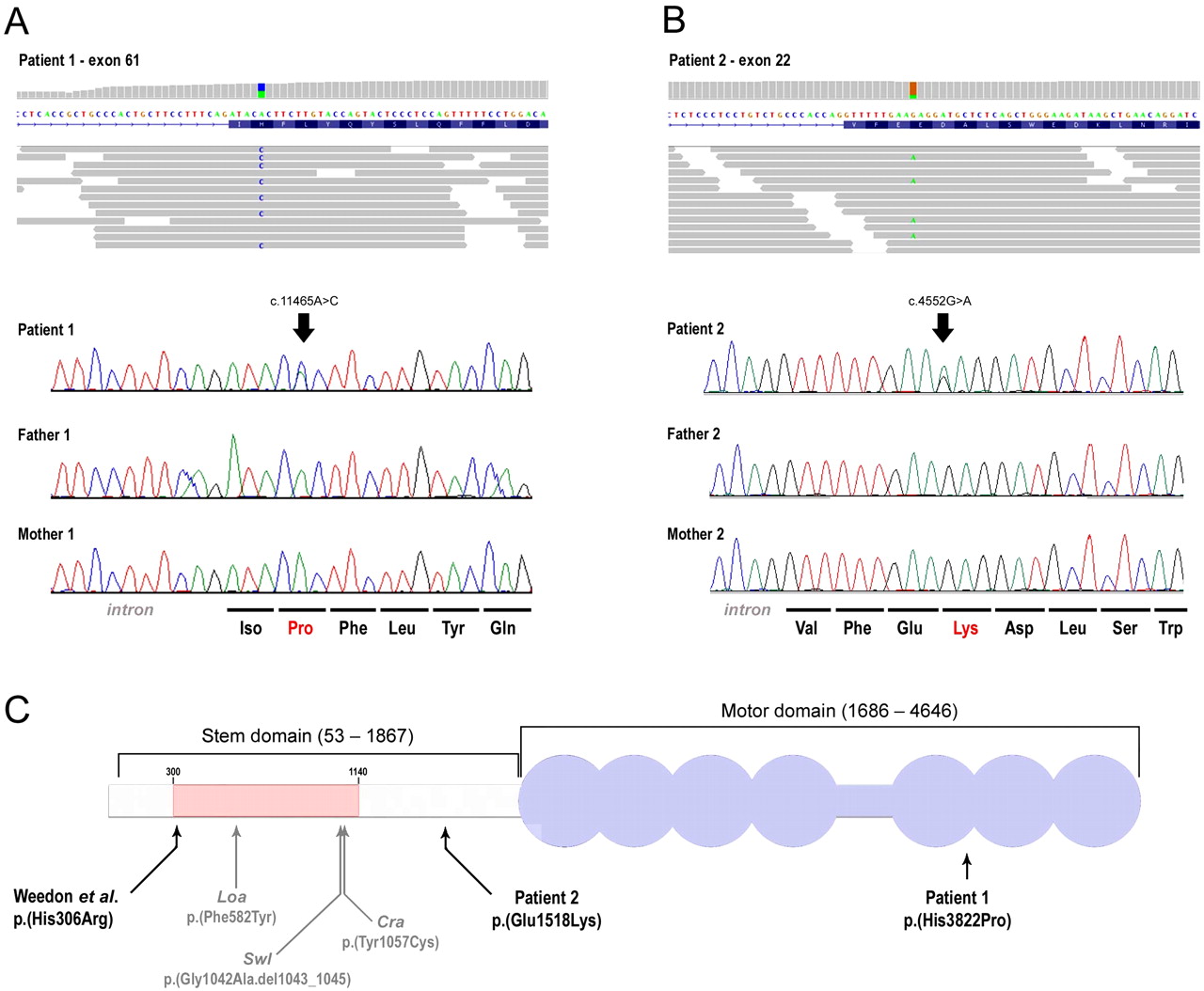
Schematic representation of human DYNC1H1 and mutations in patients 1 and 2 and the axonal (type 2) form of Charcot-Marie-Tooth (CMT2) disease family reported in Weedon et al22 together with the mutations of known mouse models. Schematic representation of the mapped exome sequencing reads visualised using the Integrative Genomics Viewer (IGV) browser for patient 1 (panel A) and patient 2 (panel B), respectively. The upper part shows the per-base coverage, with coverage represented in grey indicating the wild-type base, whereas coloured bases indicate the detection of variants. Also, a representation (part of) of exon 61 and exon 22 are provided for orientation. Individual sequence reads for patient 1 and patient 2 both show a heterozygous variant, which was followed up by Sanger sequencing, confirming the de novo occurrence in both patients. In panel C, the DYNC1H1 protein is visualised according to Weedon et al with the N-terminal region indicated by a grey horizontal bar, and the stem domain shown on above (amino acids 53–1867). The residues involving DYNC1H1 dimerisation (300–1140) are shown by a pink bar. The C-terminal motor domain (amino acids 1868–4646) is shown in purple colour, with the seven ATPase domains represented by circles and the stalk region by a horizontal bar. The equivalent positions of mutations in three mouse models, Loa, Swl, and Cra1, are shown below the representation. Note that the human protein contains two additional glycine residues at position 7 relative to mouse Dync1h1, that is, numbering of equivalent residues in human DYNC1H1 is two higher than in mouse models. The three mutations reported in the patients, including p.(His3822Pro) in patient 1, p.(Glu1518Lys) in patient 2, and p.(His306Arg) reported by Weedon et al, are indicated with arrows according to their relative positions of the functional domains of the DYNC1H1 protein.
Patient 2 was a 51-year-old woman with severe ID. She had never been able to walk or speak. She was born after an uncomplicated pregnancy with a low-normal birth weight (5th centile). She had congenital clubfeet. At the age of 3 years she developed generalised epileptic seizures. At the age of 51 years she had a height of 135 cm (<0.6th centile) and a head circumference of 52 cm (2nd centile). She had small hands and feet (<3rd centile) and the toes were short and malpositioned. Craniofacial features included brachycephaly, prominent forehead, hypertelorism, deeply set eyes, wide mouth with everted lower lip, and downturned corners of the mouth. She had secondary cataract as a result of automutilation, a kyphoscoliosis, spastic tetraplegia, and was wheelchair dependent. At the age of 50 years she had developed progressive swallowing difficulties for which a gastrostomy tube was needed.
A cerebral CT scan at the age of 46 years, performed because of persistent seizures, showed wide lateral and third ventricles, and clear signs of cortical malformation with wide opercular regions and an abnormal flat cortex with only a few, simple and shallow sulci (especially in the frontal lobes) (figure 1D,E). It was not possible to obtain a cerebral MRI scan. Additional investigations comprising a routine screen in urine and blood for metabolic disorders, including a screen for peroxisomal disorders and detailed chromosomal studies (250k single nucleotide polymorphism (SNP) array), revealed no abnormalities. A family based exome sequencing approach was also used for this patient and revealed a de novo c.4552G→A mutation in DYNC1H1, predicting a p.(Glu1518Lys) substitution (figure 2B). Similar to the mutation observed in patient 1, the Grantham score of 56 indicates a small physicochemical difference, whereas the PhyloP score at 6.03 indicates again a strong evolutionary conservation, and SIFT predicts the mutation to also have a deleterious effect for protein function. Exome coverage statistics for patients 1 and 2 and their parents are included in supplementary table 1.
Examination of 445 exomes previously sequenced in our unit for unrelated medical conditions did not show either of the changes in DYNC1H1 reported in patients 1 and 2. Moreover, in this entire cohort only 10 changes were seen in DYNC1H1 that are not present in dbSNPv132. Of these, nine variants are predicted to be synonymous changes, not affecting gene function, whereas the remaining variant predicts a missense p.Ser4603Ile substitution, which was detected in a single individual and involves a weakly conserved nucleotide (PhyloP −0.12). From this analysis, we conclude that non-synonymous changes in DYNC1H1 are very rare. Of note, retrospective analysis of genes known to cause neuronal migration disorders, including LIS1, DCX, RELN, NDE1, and TUBA1A, did not reveal any pathogenic mutations, whereas exon coverage for these genes was sufficient to detect such variants if present (supplementary table 2). Additionally, non-paternity and sample mix-up were excluded by comparison of the patients 250k Affymetrix SNP profiles to their exome data (concordance >95%, data not shown), and by the consistent segregation of uniquely inherited variants detected in the trios by exome sequencing. Thus, both changes represent bona fide de novo mutations in DYNC1H1. Combined with the fact that an average newborn is expected to have 0–2 non-synonymous de novo mutations in his or her exome, the detection of non-synonymous de novo events in DYNC1H1 in two patients with similar phenotypes strongly suggests that the mutations are causal for the phenotype.
Dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) has 78 exons and encodes the heavy chain protein (DYNC1H1) of the cytoplasmic dynein 1 motor protein complex.8 The cytoplasmic dynein 1 motor protein complex is composed of a homodimer of two heavy chains, encoded by DYNC1H1, and associated intermediate, light intermediate, and light chains. This complex is involved in several cellular functions, including spindle pole organisation and nuclear migration during mitosis, the minus end transport of cell organelles along microtubules, and retrograde axonal transport in neurons.8–11 The C-terminal region of DYNC1H1 is the motor domain of the dynein complex and is arranged as a ring with six AAA (ATPases associated with cellular activities) domains, localising ATP binding, required for energy generation, and a seventh uncharacterised domain, whereas the N-terminal region confines the stem domain, the region for homodimerisation, and binding sites for the intermediate and light intermediate chains.8 The cytoplasmic dynein 1 complex was formerly shown to play a key regulatory role in retrograde axonal transport in neurons, in which the heavy chains are responsible for movement along the microtubule.8–11 Furthermore, several studies in model organisms provided evidence that the cytoplasmic dynein 1 complex directly interacts and coprecipitates with the LIS1/PAFAH1B protein.2 3 5 12–15 The dynein heavy chain protein and the intermediate chains biochemically interact with the WD-40 repeat region of LIS1. WD-40 repeats, also known as WD or ß-transducin repeats, are short ∼40 amino acid motifs often terminating in a Trp-Asp (W-D) dipeptide, which act as a site for protein–protein interaction. Within the heavy chain, interactions with LIS1 are with the first AAA repeat, and the N-terminal cargo binding region.2 8 12
The Legs at odd angles (Loa/+), Cramping I (CraI/+), and Sprawling (Swl/+) mouse models each harbour different missense mutations in the stem domain of Dync1h1. Their phenotypes include abnormal neuronal migration and axon growth, as well as neurodegeneration, caused by defective retrograde transport.11 16–19 Studies in these three mouse models in addition showed that heterozygous missense mutations in Dync1h1 are associated with neurologic phenotypes including gait abnormalities, reduced muscle strength, reduced reflexes, and abnormal propriocepsis and nocicepsis in association with loss of motor and sensory neurons.16–19 Loa and CraI homozygous mice show a more severe phenotype and die within 24 h after birth due to inability to feed and move.17 Homozygous loss-of-function of Dync1h1 is embryonically lethal, indicating an important role of DYNC1H1 in human embryonic development.20 Interestingly, Loa mice not only exhibit defects in retrograde axonal transport, but also show clear central migration defects in hippocampus and cortex that are similar to defects observed in reduced LIS1 expression.21 The clinical findings in patients 1 and 2 strongly overlap with the phenotype observed in the Loa mouse model and strengthen the previous functional links between LIS1 and DYNC1H1.
Recently, a missense mutation c.917A→G, predicting a p.(His306Arg), was reported in a large pedigree with dominant axonal (type 2) Charcot-Marie-Tooth (CMT2) disease.22 After comparing the clinical presentation of our patients with those reported by Weedon et al,22 we noted some phenotypic overlap between patients 1 and 2 and the affected members of the CMT2 family (table 1), though the overall presentation is importantly different.
Comparison of clinical features in patients 1 and 2 and the family reported by Weedon et al22 with the axonal (type 2) form of Charcot-Marie-Tooth (CMT2) disease22
The motor abnormalities and reduced tendon reflexes that were observed in patient 1, and the congenital clubfeet in patient 2, might indicate the presence of peripheral neuropathy similar to that reported in the CMT2 family. This remains speculative, however, since neither formal neurophysiologic investigations nor peripheral nerve biopsy were performed in our patients. Of note, in the CMT2 family, four of the 13 family members with a DYNC1H1 mutation presented with global delay, speech delay and/or learning difficulties in addition to their motor problems. Whereas the delay in motor development may be explained by the peripheral neuropathy, the speech delay and/or learning difficulties obviously suggest additional involvement of the central nervous system. Brain imaging was not reported in the CMT2 family.
The important differences in phenotype between the two patients reported here and the dominant CMT2 family may indicate that heterozygous missense mutations in DYNCH1 represent a broad phenotypic spectrum. It is possible that the presentation with severe ID and neuronal migration defects depends on the location and nature of the mutation (figure 2C). The mutation in the CMT2 family is located in the stem domain at the N-terminal end of the homodimerisation region.22 In patient 1 the mutation is located in the motor domain, where no previous mutations have been documented. The mutation of patient 2 is localised in the C-terminal end of the stem domain, but outside the homodimerisation region (figure 2C). Further conclusions about genotype–phenotype correlations require that more patients with mutations in DYNC1H1 are identified and carefully characterised at the clinical level.
In summary, we conclude that de novo missense mutations in DYNC1H1 are a novel cause of severe ID associated with variable neuronal migration defects. The phenotype may include peripheral neuropathy as well.
Acknowledgments
We thank the participating patients and their parents. We also thank all technicians from the genomic disorders group for excellent technical assistance.
References
Footnotes
MHW, LELMV contributed equally to this work.
Funding This work was supported by grants from the Consortium ‘Stronger on your own feet’ to TK and MHW, The Netherlands Organization for Health Research and Development (ZonMw grants 916-86-016 to LELMV, 917-66-363 and 911-08-025 to JAV, 917-86-319 to BBAdV, and 907-00-365 to TK), the EU-funded TECHGENE project (Health-F5-2009-223143 to JdL and JAV), the AnEuploidy project (LSHG-CT-2006-37627 to BWMvB and HGB, BBAdV and JAV), the GENCODYS, an EU FP7 large-scale integrating project grant (241995 to HvB and TK).
Competing interests None.
Ethics approval Local Medical Ethical Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The article is submitted as a short report, but additional data on the exome sequencing results in these patients, including an overview of all variants detected per proband and impact of the prioritisation steps for selecting candidate non-synonymous de novo mutations, are available upon request via the corresponding author.