Article Text

Abstract
Background Joubert syndrome (JS) is a ciliopathy characterised by a distinctive brain malformation (the ‘molar tooth sign’), developmental delay, abnormal eye movements and abnormal breathing pattern. Retinal dystrophy, cystic kidney disease, liver fibrosis and polydactyly are variably present, resulting in significant phenotypic heterogeneity and overlap with other ciliopathies. JS is also genetically heterogeneous, resulting from mutations in 13 genes. These factors render clinical/molecular diagnosis and management challenging. CC2D2A mutations are a relatively common cause of JS and also cause Meckel syndrome. The clinical consequences of CC2D2A mutations in patients with JS have been incompletely reported.
Methods Subjects with JS from 209 families were evaluated to identify mutations in CC2D2A. Clinical and imaging features in subjects with CC2D2A mutations were compared with those in subjects without CC2D2A mutations and reports in the literature.
Results 10 novel CC2D2A mutations in 20 subjects were identified; a summary is provided of all published CC2D2A mutations. Subjects with CC2D2A-related JS were more likely to have ventriculomegaly (p<0.0001) and seizures (p=0.024) than subjects without CC2D2A mutations. No mutation-specific genotype–phenotype correlations could be identified, but the findings confirm the observation that mutations that cause CC2D2A-related JS are predicted to be less deleterious than mutations that cause CC2D2A-related Meckel syndrome. Missense variants in the coiled-coil and C2 domains, as well as the C-terminal region, identify these regions as important for the biological mechanisms underlying JS.
Conclusions CC2D2A testing should be prioritised in patients with JS and ventriculomegaly and/or seizures. Patients with CC2D2A-related JS should be monitored for hydrocephalus and seizures.
- JS
- ciliopathy
- CC2D2A
- seizures
- ventriculomegaly
- clinical genetics
- cell biology
- molecular genetics
- neurology
- genetics
- academic medicine
- epilepsy and seizures
Statistics from Altmetric.com
- JS
- ciliopathy
- CC2D2A
- seizures
- ventriculomegaly
- clinical genetics
- cell biology
- molecular genetics
- neurology
- genetics
- academic medicine
- epilepsy and seizures
Introduction
Joubert syndrome (JS) is a recessive disorder characterised by cerebellar vermis hypoplasia, thickened, mis-oriented superior cerebellar peduncles and an abnormally deep interpeduncular fossa resulting in the pathognomonic ‘molar tooth sign’ (MTS) on axial imaging of the mid-hind brain junction.1 2 Patients with JS manifest hypotonia, ataxia, developmental delay, abnormal eye movements and abnormal respiratory control.3 4 In addition to the core neurological features, subsets of patients with JS also display abnormalities in other organ systems, including retinal dystrophy, chorioretinal coloboma, cystic kidney disease, liver fibrosis and polydactyly.5 Given this pleiotropic phenotypic presentation, substantial overlap with other disorders is observed, including Meckel, Senior–Løken and Bardet–Biedl syndromes, as well as Leber congenital amaurosis and isolated nephronophthisis. These disorders have been classified as ciliopathies, since their causative genes all encode proteins involved in function of primary cilia, ubiquitous organelles essential in mechanical, chemical and light transduction, as well as specific signalling pathways.6
JS, like most ciliopathies, is characterised by significant genetic heterogeneity, since 13 causative genes have been identified to date: AHI1, ARL13B, CC2D2A, CEP290, INPP5E, KIF7, NPHP1, OFD1, RPGRIP1L, TCTN1, TCTN2, TMEM67 (MKS3) and TMEM216.7–20 These account individually for up to 10% and collectively for ∼50% of patients with JS, indicating that many genes are yet to be identified. In addition to the phenotypic overlap between ciliopathies, significant genetic overlap is observed, particularly between JS and Meckel syndrome (MKS), which is characterised by encephalocele, cystic/dysplastic kidney disease, ductal plate malformation of the liver, polydactyly and, typically, fetal or neonatal lethality.21 Of the 13 genes responsible for JS, six also cause MKS (CC2D2A,21 CEP290,22 RPGRIP1L,23 TCTN2,24 TMEM6712 and TMEM21614), leading to the concept that some forms of JS and MKS represent mild and severe presentations of the same biological disorder. Consistent with this concept, patients with MKS more often have truncating mutations in CC2D2A, and patients with JS more often have at least one missense mutation.21 Of note, no other genotype–phenotype correlations have been reported in patients with CC2D2A mutations.
CC2D2A mutations represent a major cause of both JS and MKS since they are responsible for ∼10% of each disorder.4 7 21 CC2D2A encodes a 1620 amino acid protein containing coiled-coiled domains and a C2 domain. Coiled-coil domains are involved in protein–protein interactions and are very common among ciliary proteins, while C2 domains are phospholipid-binding domains and have been implicated in calcium-dependent vesicle fusion and other membrane interactions.25
The present study focuses on genotype–phenotype analysis of 20 subjects with JS caused by mutations in both copies of CC2D2A. Most subjects carried at least one missense mutation, and these mutations clustered in the C2 domain and C-terminal region. Given the genetic heterogeneity of JS, correlations between the causative gene and the clinical presentation can guide diagnostic testing strategies and monitoring for complications in specific patients. Our data suggest that CC2D2A testing should be prioritised in patients with ventriculomegaly and/or seizures and patients with CC2D2A mutations should be monitored for these complications. In the future, these data will be essential for interpreting variants identified through genome-wide approaches such as whole-exome or whole-genome sequencing.26
Methods
Subjects
In cooperation with the Joubert Syndrome and Related Disorders Foundation and clinical collaborators throughout the world, the University of Washington Joubert Center has enrolled >300 families with JS under approved protocols at the University of Washington and Seattle Children's Hospital. All participants or their legal representatives provided written informed consent. Inclusion criteria were: (1) MTS on brain imaging (or cerebellar vermis hypoplasia on CT scan when an MRI was not available); (2) clinical findings of JS (intellectual impairment, hypotonia, ataxia).
Clinical and imaging data
Clinical information was collected using a structured intake form and review of medical records. Seizure status was based on documentation of seizures in medical records and/or parental report of seizures. At the time of enrolment, we reviewed brain MRI and/or CT scans to confirm the MTS and to evaluate for ventriculomegaly, agenesis of the corpus callosum, polymicrogyria, heterotopias and occipital encephalocele. When MRI or CT images were not available, we abstracted information from the MRI or CT report.
Genetic mapping and mutation identification
In consanguineous families, we first excluded known JS loci using homozygosity mapping as previously described.7 Known JS genes within heterozygous intervals in a subject were considered excluded, while genes present in homozygous intervals were sequenced. The common NPHP1 deletion was evaluated by microsatellite genotyping as previously described.27 AHI1, ARL13B, CC2D2A, CEP290, OFD1, RPGRIP1L, TCTN2, TMEM67/MKS3 and TMEM216 were sequenced in the subjects without a known genetic aetiology at the time each gene was discovered as previously described7; therefore not all genes have been sequenced in all families. Clinical genetic testing results were also included in the analysis. Segregation of mutations within a family was evaluated when parental and/or sibling DNA was available. PolyPhen-228 was used to determine the likelihood that mutations were deleterious.
In subjects harbouring a single heterozygous CC2D2A variant predicted to be deleterious, we performed array comparative genomic hybridisation to identify deletions or duplications within the CC2D2A gene. Genomic DNA was labelled and hybridised as previously described29 to a custom array with high-density probe coverage in the CC2D2A gene (average probe spacing within the gene of 41 bp).
Controls
The frequency of missense variants in subjects without severe congenital disorders was examined using data available through the NHLBI Exome Sequencing Project, Seattle, WA (URL: http://snp.gs.washington.edu/EVS/) (accessed 29 November 2011). For the p.E229del variant, we sequenced exon 9 in 96 European American control samples.
Results
Prevalence of CC2D2A mutations in JS
We identified causative CC2D2A mutations in 16 of 166 families from our JS cohort (19 individuals; table 1). To calculate the prevalence of causative CC2D2A mutations in our cohort, we used as a denominator the number of families sequenced for CC2D2A (n=166) + the number of families excluded at this locus by single-nucleotide polymorphism mapping (n=16) + the number of families not sequenced for CC2D2A because mutations in another gene had been identified as causative (n=27). Therefore the prevalence of CC2D2A-related JS in our cohort is 7.7% (16/209). In addition to these 16 families, we later enrolled one family (UW103) with CC2D2A mutations identified by clinical testing, bringing the number of subjects with CC2D2A-related JS to 20.
Summary of phenotypic information in subjects with CC2D2A mutations, single heterozygous variants, and variants of unclear functional significance identified in this cohort
Causative CC2D2A mutations
Detailed mutation analysis of all 17 CC2D2A families revealed 12 different missense mutations (five novel), eight nonsense or frameshift mutations (three novel), and two novel splice-site mutations (table 1). Missense mutations were clustered in the C2 domain (amino acids 1020–1205) and in the C-terminal region. All of these mutations were present in <1% of controls drawn from the publicly available Exome Variant Server (URL: http://snp.gs.washington.edu/EVS), and familial segregation of the mutation was consistent with recessive inheritance when parental samples were available (table 1). In 10/17 families, one missense mutation was inherited with a truncating mutation (nonsense or frameshift), while in 4/17 families, two missense mutations were found. In one family, a splice-site mutation was inherited with a frameshift mutation, and in another family a different splice-site mutation was associated with a missense mutation. Only one subject harboured a homozygous nonsense mutation. Most mutations are unique or recur infrequently between different families (figure 1), and we observed very little overlap between mutations in our cohort compared with previously published patients (figure 1 and table 2). In summary, CC2D2A mutations in JS recur rarely and generally include at least one missense mutation in the C2 domain or the C-terminal region of the protein, combined with a second missense or a truncating mutation.

CC2D2A protein schematic and variant summary. Schematic of the CC2D2A protein showing the coiled-coil domains (CC) and the C2 domain (C2). (A, B) Missense variants and in-frame deletions reported in other cohorts (A) and in our cohort (B) are indicated above the protein. (C, D) Truncating mutations (nonsense, frameshift) and splice site mutations detected in our cohort (C) and in other cohorts (D) are indicated below the protein. Novel variants from the current study are indicated (N). The number of times a specific variant was identified is indicated before the variant (n×). Variants reported in patients with Joubert syndrome are in bold. Missense variants predicted to be damaging by Polyphen-2 are black, while common variants and missense variants predicted to be benign are depicted in grey. Variants identified only as single heterozygous changes are indicated (het).
Summary of all published CC2D2A mutations, single heterozygous variants, and variants of unclear significance with phenotypic information
Heterozygous CC2D2A variants
We identified nine families with heterozygous CC2D2A variants: in seven families, only a single heterozygous CC2D2A variant was identified (one nonsense and six missense variants, of which five were predicted to be deleterious by Polyphen-2). Variants p.D1556V, p.T1116M and p.R1019X were also identified in compound heterozygosity with a second deleterious CC2D2A variant in other subjects (table 1), while variants p.R1330Q, p.K507E, p.S117R and p.L701V were found only in the heterozygous state. None of these were listed in dbSNP, and their frequency in control samples ranged from 0% to 0.8%. Deletions (or duplications) of the second CC2D2A allele were not identified by high-density custom array comparative genomic hybridisation. Between seven and eight other known JS genes were sequenced in these families without identification of the genetic cause. Two families carried a predicted damaging CC2D2A variant in the heterozygous state along with causal mutations in both alleles of another JS gene: family UW111 carried variant p.S117R in CC2D2A (Polyphen-2 score 0.507) along with two causative mutations in TCTN2 (and one predicted damaging mutation in ARL13B). Family UW54 carried variant p.K507E in CC2D2A (Polyphen-2 score 0.859) and two causative mutations in TMEM67/MKS3.
CC2D2A variants of unclear significance
In one family (UW77), we identified two missense variants that were both predicted to be benign by Polyphen-2: neither L684I nor V660I (rs16892134) are common in European American control chromosomes but occur in 0.5% and 2.4% of African American controls, respectively. Five additional JS genes were sequenced in this family with negative results. Family UW79 harboured a missense variant (c.1268G>A) downstream of a 13 bp insertion on the same allele, making it impossible to evaluate the effect of the missense variant.
In 17 families, we identified a 3 bp deletion in CC2D2A (c.685_7 delGAA) resulting in loss of a glutamic acid at position 229 (p.E229del). This variant, which is listed in dbSNP version 134 (rs112367037), was previously reported as causative for MKS, in association with a missense mutation (V1298D)32; however, we detected the p.E229del variant in 3% of control chromosomes, indicating that it is a polymorphism. Moreover, two of the families carrying this variant also had deleterious mutations in both alleles of another JS gene, explaining their phenotype. Nonetheless, it is possible that this variant has some functional effect given that two other families with the p.E229del variant (UW104 and UW105) also harboured a second CC2D2A variant strongly predicted to be damaging (IVS11(+1)G>A and D1556V), and four more families harboured single heterozygous mutations in other JS genes. In five other families, we detected another common variant (E376A, rs16892095), but this variant was present in 2.3% of combined European and African American controls.
Highly variable phenotypic presentation of CC2D2A-related JS
Phenotypic features of JS in the 20 individuals with CC2D2A-related JS were drawn from medical records, direct examination and/or family intake forms (table 1). The MTS and developmental delays/intellectual disability were present in all individuals. Alternating apnoea/tachypnoea was reported in 12/20 subjects (60%), and abnormal eye movements including nystagmus and/or oculomotor apraxia were reported in 13/19 (68%). Retinal dystrophy was found in 2/20 subjects (10%), kidney disease in 4/19 (21%) and liver disease in 2/18 (11%), with one subject requiring a kidney transplant and another requiring a liver transplant. The subject with severe liver disease requiring transplantation also had kidney disease and chorioretinal coloboma and was previously reported to have a diagnosis of COACH syndrome.30 No subjects with CC2D2A mutations had polydactyly. Although these frequencies of organ involvement are somewhat lower than what is generally reported in the literature for JS,4 5 34 the number of subjects is too small to draw definitive conclusions.
Enrichment for abnormal neuroimaging findings, in particular ventriculomegaly, in CC2D2A-related JS
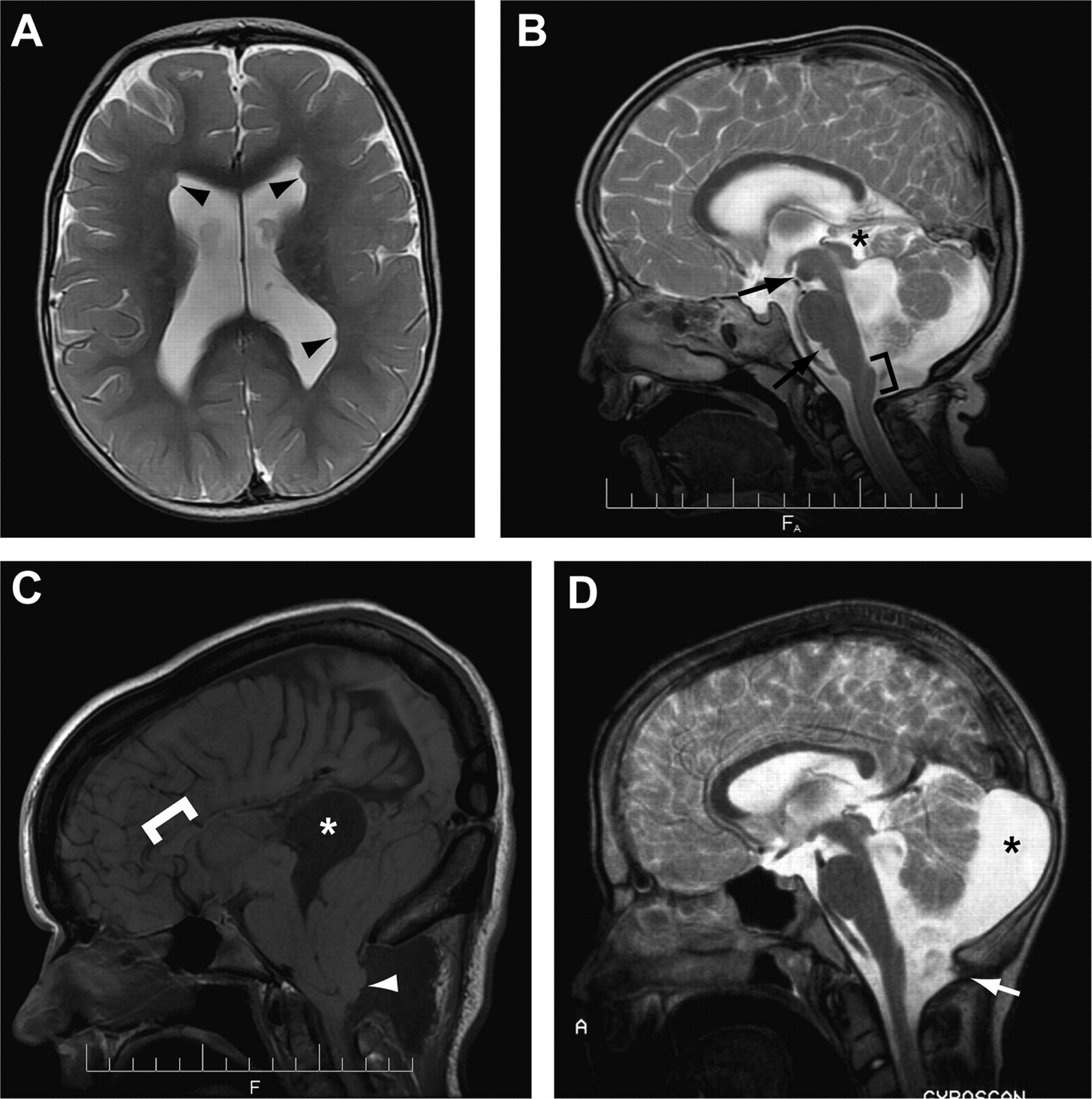
In addition to the MTS, a variety of abnormal neuroimaging features are rarely reported in patients with JS.35 We reviewed full brain MRI studies from 13 of our 20 subjects with CC2D2A-related JS, and identified a variety of additional neuroimaging abnormalities (table 3 and figure 2). Based on the imaging review and information from medical records, 13/17 (76%) subjects with CC2D2A mutations had ventriculomegaly (figure 2A), compared with 21/105 (20%) subjects without CC2D2A mutations (p<0.0001, Fisher exact test). Three subjects had hydrocephalus requiring ventriculoperitoneal shunting, and one subject without ventriculomegaly (UW47-3) had a small but clear increase in ventricle size between 3 and 11 years of age (based on imaging at these ages).
Neuroimaging features in subjects with CC2D2A-related JS

Selected atypical neuroimaging features in subjects with CC2D2A-related Joubert syndrome. (A) Ventriculomegaly and multiple subependymal heterotopias (black arrowheads) in UW103-3. (B) Interpeduncular and infrapontine heterotopias (black arrows), dysplastic tectum (asterisk) and abnormal prominence of the cervicomedullary junction (bracket) in UW82-3. (C) Agenesis of the posterior corpus callosum (bracket indicates remaining corpus callosum) and severe inferior cerebellar ectopia (white arrowhead) in UW49-3. Note the 4th ventricular cyst herniating superiorly through the tentorium (area of low signal around the asterisk) and the inferior pseudomeningocele due to prior posterior fossa decompression surgery (area of low signal around the white arrowhead). (D) Retrocerebellar fluid collection (asterisk) and foramen magnum cephalocele (white arrow) in UW79-4. The “A” is on the original MRI image and refers to Anterior. MRI sequences are as follows: (A) axial T2-weighted, (B and D) sagittal T2-weighted and (C) sagittal T1-weighted.
Additional infratentorial abnormalities included asymmetrical cerebral peduncles (n=2), occipital cephalocele (n=1), posterior fossa fluid collections (two retrocerebellar (figure 2D), and one originating in the 4th ventricle and herniating through the tentorium (figure 2C)), heterotopias (two interpeduncular, one inferior to the pons (figure 2B), one cerebral peduncle), foramen magnum cephalocele (n=2), and abnormal dorsal prominence at the cervicomedullary junction (n=2). Additional supratentorial abnormalities included agenesis and/or dysgenesis of the corpus callosum (one subject with complete agenesis, two with posterior agenesis (figure 2C), four with global thinning), polymicrogyria (one with seizures, one without seizures), cortical heterotopias (two subependymal (figure 2A), one transmantle), increased T2/FLAIR signal in the white matter (n=3), and malformed hippocampi (n=1). Three subjects also had striking dolichocephaly. In addition, fetal siblings of UW50-3 and UW103-3 were reported to have encephaloceles. In summary, ventriculomegaly and other neuroimaging findings rarely observed in patients with JS were substantially enriched in subjects with CC2D2A-related JS.
Enrichment for seizures in CC2D2A-related JS
In addition to the striking enrichment of ventriculomegaly in subjects with CC2D2A-related JS, we noted that seizures, also a rare finding in patients with JS, were reported in 5/17 families with CC2D2A mutations (27%) (figure 3A). Seizure status in all subjects was based on information available at the time the subject was enrolled (parental report and/or medical records). To reduce potential bias due to selective inclusion of subjects with CC2D2A mutations, we repeated the analysis using only the families enrolled at the time we sequenced the entire cohort for CC2D2A and for whom CC2D2A status had been clarified (by sequencing (n=166), mapping (n=16) or if another causative gene was identified (n=27) for a total of 209 subjects with known CC2D2A status). In this analysis, seizures were present in 5/16 families with CC2D2A mutations while they were reported in 17/193 families without CC2D2A mutations (p=0.0163, Fisher's exact test).

{kind=link}
{kind=link}
{kind=link}
Seizures in Joubert syndrome (JS). (A) Pie chart indicating the relative contributions of different genes in subjects with seizures. Absolute numbers are indicated for each gene. ‘Unknown aetiology’ indicates that the underlying genetic cause has not been identified. (B) Bar graph showing the different types of seizures in subjects with JS. Black bars indicate CC2D2A subjects, and white bars indicate the rest of the subjects with seizures. ‘Unknown’ indicates that the seizure type could not be determined.
To gain additional information on seizure type, age of onset and severity, we sent a survey to all subjects with reported seizures (survey available upon request). Sixteen of 22 surveys were returned, which, in combination with the previously available records, enabled classification of the seizures for 19 of 22 subjects (figure 3B). In the remaining three subjects, we were unable to obtain any additional information beyond the initial report of seizures (classified as ‘unknown’ seizure type in figure 3B). We observed a variety of seizure types among the 22 patients with seizures, regardless of the genotype. In the CC2D2A subgroup, we observed complex partial seizures in two subjects, primary generalised seizures in one subject, and unknown seizure type in one subject (table 4). In non-CC2D2A subjects, absence seizures and simple partial seizures were also reported. After analysis of survey responses, several subjects did not meet criteria for a seizure disorder: three subjects had seizures exclusively in the neonatal period or in infancy and were currently seizure-free off medication; two other subjects had clearly provoked seizures: one (without CC2D2A mutations) had febrile seizures and the second (with CC2D2A mutations) had a single seizure immediately after liver transplant, thought to be secondary to high ciclosporin levels. Finally, the diagnosis of seizures was considered questionable in two patients, where no formal diagnosis had been made and where the parental description appeared ambiguous. On the basis of this additional information, a seizure disorder was present in 4/16 families with CC2D2A mutations and 12/193 families without CC2D2A mutations (p=0.024, Fisher exact test), while six subjects (one with CC2D2A mutations) were excluded because they only had neonatal or provoked seizures.
Neurological features and neurodevelopmental outcome of CC2D2A- related Joubert syndrome
Of the 16 subjects with a bona fide seizure disorder and known CC2D2A mutation status, four had definitive CC2D2A mutations, one had compound heterozygous CC2D2A variants of unknown significance (UW77-3), one each had causal mutations in TMEM216, NPHP1 and AHI1, and eight did not have a known cause (figure 3A).
Neurological and neurodevelopmental outcome in CC2D2A-related JS
Since subjects with JS due to CC2D2A mutations have a higher prevalence of seizures, ventriculomegaly and other neuroimaging findings, we further investigated their neurological and neurodevelopmental outcomes. For this purpose, we sent a basic developmental survey to all 17 families with CC2D2A mutations, leading to 13 responses (survey available upon request). We found a wide range of developmental outcomes, similar to previous descriptions of outcomes in patients with JS (table 4). Independent walking was achieved in 8/17 subjects (age range 2–9 years). A surprisingly high number of parents reported involuntary movements (5/20 subjects) and tics (3/20 subjects). Verbal communication was achieved in 7/15 subjects, but two of these seven subjects also used sign language. Sign language was the main mode of communication for 4/15 subjects, and an augmentative communication device or a picture communication system was used in 3/14. One subject was non-verbal, but no additional information about other modes of communication was available. All subjects for whom we have educational information were enrolled in special education, with the highest functioning individual attending post-secondary school. Behavioural problems reported by the families included aggression/violence (n=4), temper outbursts (n=1), anxiety (n=1) and autism spectrum disorder (n=2). Additional neurological problems reported included recurrent facial palsy, cortical blindness and hemifacial spasms (table 4). In summary, the increased prevalence of ventriculomegaly and seizures in CC2D2A-related JS does not appear to be associated with a markedly increased frequency/severity of other neurological features or an obviously poorer developmental outcome compared with published reports.4 36–38
Discussion
Mutations in CC2D2A account for 7.7% of JS in our large cohort and for 10% of MKS,21 highlighting the importance of this gene in human ciliopathies. As a consequence, genotype–phenotype correlations for CC2D2A mutations are of direct use in patient care, especially for JS, since genotype–phenotype correlations can guide genetic testing, optimise monitoring for medical complications and, ideally, provide better prognostic information for newly diagnosed patients. In the future, genotype–phenotype information will be critical for the interpretation of variants identified by genome-wide sequencing technologies that will soon make their way into clinical use.
Genotype–phenotype correlations
Our work identifies two neurological features uncommon in JS, namely ventriculomegaly and seizures, as significantly enriched in subjects with CC2D2A-related JS. In particular, ventriculomegaly was much more common in subjects with CC2D2A-related JS compared with our other subjects and previously published patients.35 At least some of the ventriculomegaly is due to hydrocephalus (in three subjects), but we also have evidence for very slow volume loss on serial MRIs in one subject. Given the small sample size, the associations between CC2D2A mutations and ventriculomegaly/seizures will need to be validated in other JS cohorts; however, our findings indicate that genetic testing should be preferentially directed towards CC2D2A sequencing in patients with ventriculomegaly/hydrocephalus and/or seizures. Conversely, the threshold for seizure work-up and neurological consultation should be low when seizures are suspected in patients with CC2D2A-related JS. Despite the more prevalent neuroimaging abnormalities and seizures in CC2D2A-related JS, these subjects did not have markedly more severe neurodevelopmental outcomes, nor did they have more common involvement of the retina, kidneys and liver. The one possible exception to this conclusion is the recurrent reports of involuntary movements and tics, which are inconsistently reported in patients with JS.
Limitations
This study has several limitations that are typical for research on the natural history of rare disorders. Although we report the largest number of subjects with CC2D2A-related JS to date, the numbers remain small. In addition, known and unknown ascertainment biases are likely. For instance, families of children with more severe disease may be more likely to enrol in research studies. Furthermore, we are not able to perform standardised assessments on subjects who live at a distance and therefore may misclassify subjects with respect to clinical features. The progressive retinal, kidney and liver complications may also be underascertained in younger patients. To improve on the information in this report, future studies would have to use more standard assessments on larger numbers of subjects.
Implications for CC2D2A function
Analysis of the CC2D2A mutations in our JS cohort suggests a number of points relevant for CC2D2A protein function. C2 domains are phospholipid-binding domains that are often involved in vesicle trafficking and fusion. Recent evidence implicates Cc2d2a in Rab8-dependent vesicle trafficking in zebrafish photoreceptors,39 similar to what has been described for other JS proteins such as AHI1.40 Clustering of mutations in the C2 domain is consistent with a role for CC2D2A in vesicle trafficking, and a role for aberrant trafficking in the molecular mechanism underlying CC2D2A-related JS. Multiple mutations are also present in the C-terminal portion of the CC2D2A protein, which does not contain defined functional domains, but shares similarity with centrosomal protein CEP76.7 Indeed, a BLAST search with the C-terminal CC2D2A sequence (amino acids 1205–1620) retrieves CEP76 and vice versa. This finding suggests the presence of an as yet unidentified functional domain.
In addition, we confirm the previous finding that patients with JS harbour at least one missense mutation in CC2D2A, since only one of our 20 subjects had a homozygous nonsense mutation, while all others had either two missense mutations or one missense and one truncating mutation. Moreover, several truncating mutations in our JS subjects are located in the C-terminal region of the protein, suggesting that at least some partially functional protein might be produced. This contrasts with the finding that patients with MKS have two truncating mutations, and that the majority of these are within or before the C2 domain, indicating that functional protein is unlikely to be produced.21 Therefore patients with CC2D2A-related JS are likely to have some residual CC2D2A protein function, whereas patients with MKS are more likely to have severe loss-of-function or complete absence of CC2D2A.
Impact of genetic modifiers on phenotype
Reliable genotype–phenotype correlations are rare in the clinically and genetically heterogeneous ciliopathies. Nevertheless, causative mutations in some genes appear to be more highly associated with specific features, as highlighted by the association of TMEM67 mutations with liver involvement41 and of CC2D2A mutations with ventriculomegaly and seizures (present study). Moreover, the severity of the causative mutations correlates at least in some cases with the severity of the phenotypic presentation (CC2D2A mutations leading to MKS vs JS). Beyond these associations, major phenotypic variability is observed. This could be explained by a prominent hypothesis in the field that proposes that the phenotype in a given patient results from the sum of the effects of variants in ciliary (and other) genes, also termed ‘mutational load’.42 Support for this hypothesis comes from Bardet–Biedl syndrome, which in some instances exhibits oligogenic inheritance.43–45 For JS and its allelic disorders (ie, nephronophthisis, Leber congenital amaurosis, Senior–Løken syndrome and MKS), specific variants in RPGRIP1L and AHI1 have been proposed to act as genetic modifiers of retinal disease risk.46 47 Similarly, MKS1 variants have been proposed to be associated with seizures in patients with Bardet–Biedl syndrome.48 Although we lack the statistical power to determine their significance, we observed several intriguing combinations of heterozygous variants in multiple JS genes within individual subjects. Taking all these data together, it is likely that the effects of mutations in a causal gene drive much of the phenotype in a given patient, while variants in modifier genes explain a substantial portion of the phenotypic variability between patients with mutations in the same causal gene. Integrating comprehensive sequence variant data with reliable phenotypic information to develop predictive models for phenotype, as well as identifying the highest-yield molecular targets for therapeutic interventions, remain challenges for future research.
Acknowledgments
We would like to thank all participating families with Joubert syndrome and the Joubert Syndrome and Related Disorders Foundation.
References
Footnotes
Funding This work was supported by National Institute of Health grants 5KL2RR025015 and R01NS064077 to DD, and K12HD043376 to RB-G, by Basil O'Connor Start Scholar Research Grant No 5-FY09-13 from the March of Dimes Foundation to DD, and by an Arc of Washington Trust Fund grant to DD. We would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by University of Washington IRB and Seattle Children's Hospital IRB.
Provenance and peer review Not commissioned; externally peer reviewed.