Article Text

Abstract
Background Nephronophthisis associated ciliopathies (NPHP-AC) comprise a group of autosomal recessive cystic kidney diseases that includes nephronophthisis (NPHP), Senior-Loken syndrome (SLS), Joubert syndrome (JBTS), and Meckel-Gruber syndrome (MKS). To date, causative mutations in NPHP-AC have been described for 18 different genes, rendering mutation analysis tedious and expensive. To overcome the broad genetic locus heterogeneity, a strategy of DNA pooling with consecutive massively parallel resequencing (MPR) was devised.
Methods In 120 patients with severe NPHP-AC phenotypes, five pools of genomic DNA with 24 patients each were prepared which were used as templates in order to PCR amplify all 376 exons of 18 NPHP-AC genes (NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290, GLIS2, RPGRIP1L, NEK8, TMEM67, INPP5E, TMEM216, AHI1, ARL13B, CC2D2A, TTC21B, MKS1, and XPNPEP3). PCR products were then subjected to MPR on an Illumina Genome-Analyser and mutations were subsequently assigned to their respective mutation carrier via CEL I endonuclease based heteroduplex screening and confirmed by Sanger sequencing.
Results For proof of principle, DNA from patients with known mutations was used and detection of 22 out of 24 different alleles (92% sensitivity) was demonstrated. MPR led to the molecular diagnosis in 30/120 patients (25%) and 54 pathogenic mutations (27 novel) were identified in seven different NPHP-AC genes. Additionally, in 24 patients only single heterozygous variants of unknown significance were found.
Conclusions The combined approach of DNA pooling followed by MPR strongly facilitates mutation analysis in broadly heterogeneous single gene disorders. The lack of mutations in 75% of patients in this cohort indicates further extensive heterogeneity in NPHP-AC.
- Next-generation sequencing
- ciliopathy
- nephronophthisis
- genetics
- renal medicine
Statistics from Altmetric.com
Introduction
Dysfunction of the primary cilium/basal body complex has been implicated in the pathogenesis of nephronophthisis associated ciliopathies (NPHP-AC), including nephronophthisis (NPHP), Senior Loken syndrome (SLSN), Joubert syndrome (JBTS), and Meckel–Gruber syndrome (MKS).1 NPHP-AC are rare, genetically heterogeneous, autosomal recessive inherited disorders, which share a broad phenotypic spectrum as a result of ciliary/centrosomal defects in various cell types—for example, retinal photoreceptors or renal tubular epithelial cells.2 In NPHP, kidney tubular basement membrane disintegration, tubular atrophy, interstitial fibrosis, and cyst formation result in progressive renal failure during childhood or adolescence. About 15% of patients develop extrarenal manifestations, in particular, progressive retinal dystrophy, referred to as SLSN. In patients with JBTS, midbrain–hindbrain malformations and cerebellar vermis hypoplasia/aplasia result in numerous neurological features including developmental delay, intellectual disability, muscle hypotonia, ataxia, oculomotor apraxia, nystagmus, and irregular breathing patterns in neonates.3 The most severe manifestation of the NPHP-AC clinical spectrum is seen in fetuses with MKS, a perinatally lethal ciliopathy, characterised by central nervous system malformations (typically occipital encephalocoele), bilateral postaxial hexadactyly, hepatobiliary ductal plate malformation, and multicystic dysplastic kidneys. Mutations in 18 different recessive genes have been identified as the molecular cause in NPHP-AC (table 1). Twelve genes have been implicated in NPHP and/or SLSN (NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290, GLIS2, RPGRIP1L, NEK8, TMEM67, TTC21B, and XPNPEP3).4–15 Ten are known to cause JBTS (AHI1, TMEM216, INPP5E, NPHP1, CEP290, RPGRIP1L, TMEM67, ARL13B, CC2D2A, TTC21B) (Davis et al, submitted, 2010).16–24 Mutations in five genes (MKS1, TMEM67, CEP290, RPGRIP1L, CC2D2A, TMEM216) have been shown to cause MKS.12 17 25–28 Multiple allelism within the NPHP-AC phenotypic spectrum has been recurrently reported for many of these genes, especially CEP290, RPGRIP1L, TMEM67, CC2D2A, TTC21B, and TMEM216. For example, hypomorphic missense mutations in the gene TMEM67 (MKS3/NPHP11) are implicated in NPHP with liver fibrosis and JBTS type 6, whereas truncating mutations in TMEM67/MKS3 have been reported in MKS cases with severe developmental and dysplastic phenotypes.14 22 26 The presence of multiple allelism and broad heterogeneity together with extensive phenotypic clinical overlap in patients with NPHP-AC requires extensive mutational analysis efforts in order to identify the underlying molecular aetiology. The challenge of analysing increasing numbers of candidate genes associated with disease in large cohorts of patients can now be met by applying next generation sequencing technologies. We chose to perform single PCR reactions with well established primer pairs, rather than trying to implement multiplexing, in order to amplify all coding exons (376) of 18 known NPHP-AC disease genes. In order to reduce the calculated number of 45 120 PCR reactions, required to amplify all 376 exons in 120 individuals, we devised a pooling strategy by generating five DNA pools derived from 24 individuals each. Identification of the mutation carrying individuals was done by CEL1 endonuclease based heteroduplex analysis and Sanger sequencing.29 We demonstrate that the approach of pooling DNA samples in combination with massively parallel resequencing (MPR) is a robust, economic, and highly effective method for examining larger patient cohorts for mutations, especially in diseases with broad genetic locus heterogeneity.
Genes investigated by massively parallel resequencing in 120 patients with severe nephronophthisis associated ciliopathies (NPHP-AC)
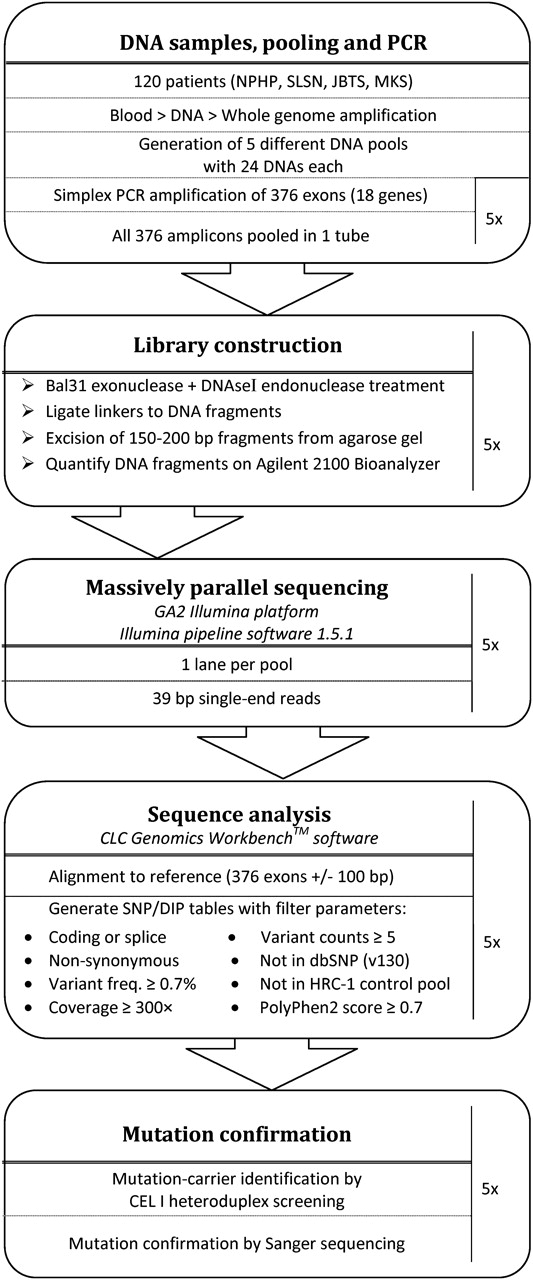
An overview of the various steps undertaken, including DNA pooling, PCR, MPR, and mutation carrier identification is depicted in a flowchart (figure 1).

Flowchart illustrating the various steps of the DNA pooling and massive parallel resequencing approach, which was applied in order to perform mutation analysis for 18 genes in 120 patients with nephronophthisis associated ciliopathies. JBTS, Joubert syndrome; MKS, Meckel–Gruber syndrome; NPHP, nephronophthisis; SLSN, Senior Loken syndrome.
Patients and methods
Human subjects
We obtained blood samples, pedigrees, and clinical information after receiving informed consent (http://www.renalgenes.org) from all patients. Approval for experiments on humans was obtained from the University of Michigan Institutional Review Board. The cohort of 120 patients with severe NPHP-AC included 54 (45%) patients diagnosed as JBTS with kidney involvement, 14 (12%) patients with SLSN, six (5%) patients with MKS, and 46 (38%) patients with NPHP and early onset of end stage renal disease before age 7 years. The cohort consisted of 56 (47%) familial cases versus 64 (53%) sporadic cases. Consanguinity was known to be present in 30 (25%) families. As a first diagnostic step, homozygous deletions of NPHP1 were excluded in all patients by multiplex PCR.29 Total genome homozygosity mapping was performed in 114 of 120 patients. In patients exhibiting long runs of homozygosity (most likely by descent) at known NPHP-AC loci, exon sequencing for respective genes was carried out and was negative. Ninety-six healthy control DNA samples (Human Random Control DNA panel-1, HRC-1) were obtained from the European Collection of Cell Cultures (ECACC, Salisbury, UK).
Whole genome amplification and DNA pooling
In order to normalise various DNA samples of dissimilar quality, whole genome amplification (WGA) was performed by individually amplifying 10 ng genomic DNA of 120 different individuals and 96 healthy control samples using Phi29 based DNA polymerase strand displacement amplification according to the manufacturer's instructions (GenomiPhi DNA amplification kit, GE Healthcare, Buckinghamshire, UK). Subsequently, DNA was purified using 96-well spin columns (Rapid 96TM PCR purification system, Marligen-Biosciences, Ijamsville, MD, USA). Genomic DNA of 24 individuals was pooled at 2 μg each and diluted to 60 ng/μl. Five pools were generated to represent 120 individuals. As a control, another equimolar DNA pool was generated, by pooling 96 DNA samples derived from healthy individuals of Caucasian origin (Human Random Control DNA Panel-1 (HRC-1)).
PCR amplification
All 376 exons of the genes NPHP1 (20 exons), INVS (16), NPHP3 (28), NPHP4 (30), IQCB1 (15), CEP290 (54), GLIS2 (6), RPGRIP1L (27), NEK8 (15), TMEM67 (28), INPP5E (10), TMEM216 (5), AHI1 (27), ARL13B (10), CC2D2A (37), TTC21B (29), MKS1 (18), and XPNPEP3 (10) were individually amplified using pairs of exon flanking primers (supplementary table 1) in each of the DNA pools as the PCR template. A 12 μl total volume single PCR reaction was set up with 120 ng (2 μl) of pooled DNA derived from 24 different patients (5 ng of each DNA), 2.5 pmol of each forward and reverse primer, and 6 μl HotStar-Taq Polymerase mixture (Qiagen, Hilden, Germany). DNA amplification was performed on a thermal cycler (Mastercycler, Eppendorf) using Thermo-Fast 96-well plates (ABgene), applying the following touchdown PCR protocol for all PCR reactions: initial denaturation at 94°C for 15 min, followed by 24 cycles with an annealing temperature decreasing 0.7°C per cycle, starting at 72°C for 30 s; denaturation at 94°C for 30 s, and extension at 72°C for 1 min. An additional 20 cycles with fixed annealing temperature were added: 94°C for 30 s, 55°C for 30 s, 72°C for 1 min with a final extension of 72°C for 10 min. Two microlitres of each reaction was analysed by agarose gel electrophoresis (1.2% agarose, 120 V, 1 h).
Enzymatic modification of PCR products before MPR
For each pool, we combined 5 μl of each of the 376 exonic PCR products ranging from 139 bp to 1236 bp and purified the DNA fragments on three separate columns of a QIAquick PCR Purification Kit (Qiagen). In order to generate random start positions for next generation sequencing, to reduce the presence of primer sequences and adjacent intronic sequences, and to reduce fragment sizes, we digested the mixture of PCR products with BAL-31 exonuclease and DNase I endonuclease. Ten micrograms of the purified PCR fragment mixture was digested with 5 units of BAL-31 exonuclease (New England Biolabs, Ipswich, MA, USA) for 5 min at 30°C in a 200 μl reaction. The reaction was stopped by adding EGTA to a final concentration of 20 mM and immediately heat inactivated at 65°C for 10 min, followed by Qiaquick PCR column purification (Qiagen). Remaining DNA (about 2 μg) was further digested by incubating with 1 unit DNAse I (Roche, Basel, Switzerland) and freshly prepared reaction buffer (2× reaction buffer contains 20 mM Tris-Cl (pH 7.5), 2 mM CaCl2, and 20 mM MnCl2) for 3 min at 16°C. In the presence of Mn2+ ions, the DNase I enzyme cleaves both DNA strands at approximately the same site.30 The reaction was stopped by adding 2 μl EDTA (500 mM) and immediately heat inactivated at 65°C for 10 min. Afterwards, DNA was purified using Qiaquick columns (Qiagen) and eluted with 30 μl EB buffer.
Next generation sequencing on a Solexa/Illumina GA2 platform
Library construction of the modified PCR fragment mixture was performed using ‘Genomic DNA Sample Prep Kit’ according to the manufacturer's instructions (Illumina, San Diego, California, USA). Fragments were separated on a 1.5% agarose gel and excised in the 150–200 bp range. Fragments were then purified and subjected to 10 rounds of PCR amplification using complementary linker specific primers. The amount and size distribution of each sample was analysed on a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). For next generation sequencing, single stranded DNA fragments were annealed to a flow cell surface in a cluster station (Illumina) and subjected to 46 cycles of bridge amplification. Fragments were run on a single lane of a Solexa/Illumina Genome Analyser II platform, generating about 15–20 million single-end sequence reads of 39 bases each. Image analysis and base calling were generated by the Genome Analyser Pipeline 1.5.1 with default parameters. Illumina specific FASTQ files containing sequence information and quality scores for each base call were exported for downstream analyses.
Sequence read mapping and variant calling
Sequence alignment was performed with ‘CLC Genomics Workbench’ software (CLC-bio, Aarhus, Denmark) using imported and annotated human reference genome assembly hg18 (NCBI build 36) chromosome sequence files as a reference. We annotated all 24 human chromosomal reference datasets with gene transfer files (GTF) after downloading dbSNP(v130) from UCSC and by annotating all obligatory splice sites. After importing the concatenated FASTQ (Illumina) files generated by the GAII pipeline into the CLC software, sequence reads were mapped to exonic coding regions plus adjacent 100 bp intronic sequence of all 376 initially PCR amplified targets. Gapped alignments with default parameters applying bioinformatic costs for mismatches of ‘2’ and indel costs of ‘3’ were performed. Variant calls were obtained using the following filter parameters: coverage ≥300X, variant frequency of at least 0.7%, and a minimum variant count of five reads. The variant analysis included coordinates of obligatory splice sites, and all variants predicted to change the amino acid sequence (missense, nonsense, and coding indels). When calling variants from normal reference sequence (VRS), we excluded all synonymous variants and all variants localised in non-coding exonic regions or in introns (except obligatory splice sites). Furthermore, we excluded known single nucleotide polymorphisms (SNPs) (v130) and all variants identified in the healthy control pool MPR experiment processed in parallel using identical filter parameters (coverage, minor allele frequency, and minimal counts). To prioritise for downstream analysis we scored missense variants according to the information of evolutionary conservation and the likelihood of a potential protein damaging effect using PolyPhen2 software predictions (http://genetics.bwh.harvard.edu/pph/).31 All variants with a predicted ‘probable’ or ‘possible’ damaging effect and a score above 0.7 (PolyPhen2) were further analysed in the original unpooled DNA samples of 24 pooled individuals by CEL1 heteroduplex analysis and/or direct Sanger sequencing.
Identifying mutation carriers out of 24 pooled individuals
In order to identify the carrier(s) of the selected VRS (114 altogether) from a pool of 24 patients, standard mutation detection techniques were applied. The mutation containing exon was amplified by PCR using genomic DNA of all 24 patients as template in separate reactions. Subsequently, the PCR products were analysed using CEL I heteroduplex screening or using direct Sanger sequencing.29 In all cases in which only one mutated allele was discovered, we screened for a potential second mutated allele by Sanger sequencing all exons of the respective gene.
Results
Proof of principle for pooled DNA sample analysis
In a pilot project we tested the DNA pooling and next generation sequencing strategy by pooling the DNA of 18 patients (36 alleles) with known mutations in 18 different NPHP-AC causing genes (table 1, supplementary table 2). The equimolar DNA pool contained altogether 14 different heterozygous and 10 different homozygous mutations/variants. From a total of 24 different mutations/variants, nine were missense, eight were nonsense, two were small insertion/deletions, two were synonymous rare polymorphisms, and three were affecting obligatory splice sites. After MPR of 376 different pooled PCR products derived from the DNA pool of 18 different mutation carriers, the analysis revealed confirmation of 22 out of 24 known mutations/variants (92%) (supplementary table 2). For the two mutations (p.S360T in NPHP3, and R85X in TMEM216) that were not detected, there was a lack of coverage of the respective exons with only two or zero variant sequence reads, respectively (supplementary table 2). Mutations were detected after filtering for variants affecting amino acid residues, reading frames, or obligatory splice sites by setting parameters for variant calls for minor allele frequency to ≥0.7%, minimum coverage to ≥300×, and the minimum number of reads (counts) to ≥5 as described in methods.
Statistics on Solexa/Illumina massively parallel resequencing runs of five DNA pools derived from 120 patients with nephronophthisis associated ciliopathies (NPHP-AC)
Statistics on Solexa/Illumina GA2 sequence runs of pooled DNA samples
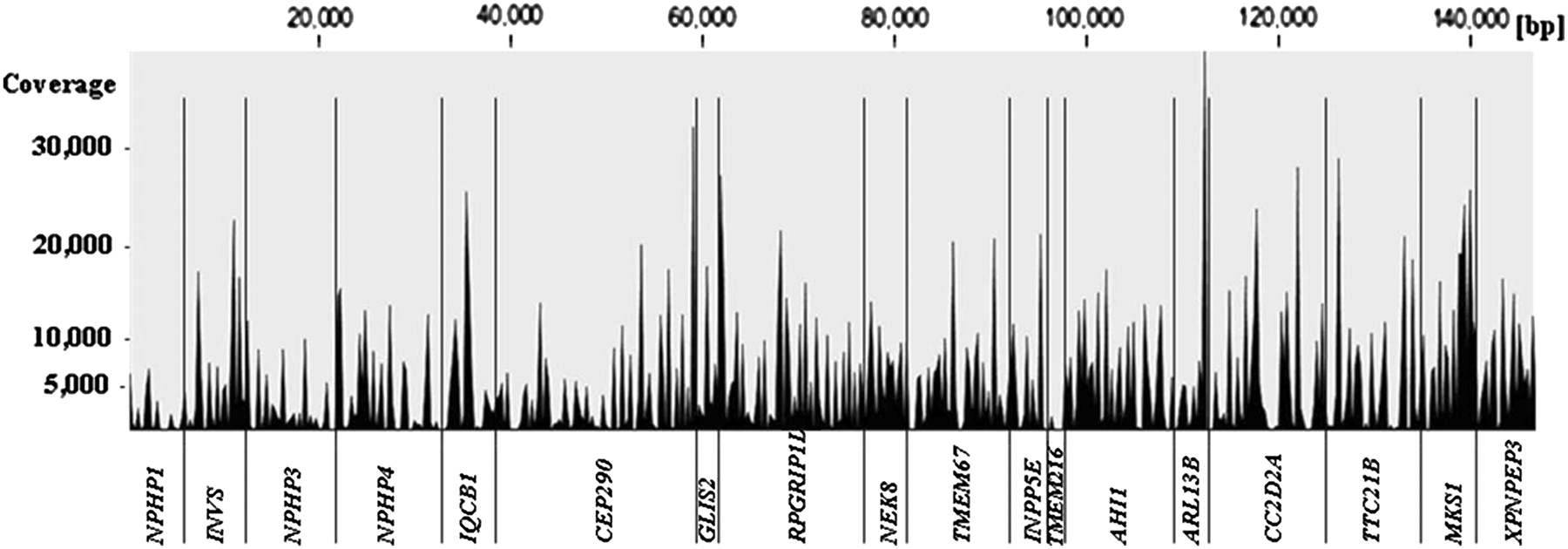
Next generation sequencing of 376 different PCR products generated by using pooled DNA samples was carried out on an Illumina/Solexa GA2 platform (one pool per lane of a flow cell). In addition to five experimental patients' DNA pools (patient-pool 1>5) generated by pooling 24 samples each, we sequenced the PCR products derived from a pool of 96 healthy control individuals (HRC1-individuals) and one positive control pool derived from 18 patients with known mutations. Statistical features of Solexa/Illumina sequence runs representing the five experimental patient pools are shown in table 2. One lane of am eight lane flow cell GA2 run delivered on average about 16.7 million (range 15.5–19.9 million) short reads of 39 bases. After gapped alignment to the human reference sequence, 85% of all reads mapped back to one of the 376 targeted amplicons (18 genes). The sequence concatenation of all 376 amplicons amounts to a total length of 147.5 kb, 52.8 kb of which were exonic coding regions. The median coverage for coding nucleotide sites was 4186X (mean 5753X), which translates into a median depth of 87X (mean 120X) per site per single allele, respectively. An example of the overall coverage of all concatenated exons derived from 18 NPHP-AC genes is shown in figure 2 for patient pool 3. The overall maximum coverage of 54 386X was found in patient pool 5. On average, about 95% of the targeted coding regions were sufficiently covered at least 300-fold in order to reliably call a heterozygous mutation/variant present on only one out of 48 pooled alleles derived from 24 patients.

Obtained coverage depth along the entire concatenated sequence of all 376 amplicons (145 kb) derived from 18 different nephronophthisis associated ciliopathies (NPHP-AC) genes. Shown as an example are the results from the massively parallel resequencing (MPR) mutation analysis performed on one lane of a Solexa/Illumina flowcell in patient pool #3 (DNA pool of 24 patients). Note that all exonic and adjacent intronic regions of all amplicons are shown. The median coverage depth within exonic coding regions (total of 54 kb) is 4186×. About 95% of all coding bases are covered at least 300-fold.
Mutation carrier identification by CEL I endonuclease heteroduplex screening and Sanger sequencing
MPR of PCR products of 120 individuals revealed initially a total of 379 variants within coding regions of 18 genes analysed, 119 of which were known SNPs (table 2). Performing MPR on a healthy control sample pool in parallel reduced the remaining 260 variants by another 49 variants (table 2). These variants were either unknown SNPs identified in the human random control pool or false calls due to software base calling or alignment artefacts. Of the remaining 211 variants, 114 truncating mutations (nonsense, frameshift), canonical splice sites and missense changes with a predicted ‘damaging’ impact at the protein level (PolyPhen2 score >0.7) were followed up by identification of the contributing individual. CEL I heteroduplex screening and direct Sanger sequencing were subsequently performed for the 114 variants in order to identify the mutation carrier(s) out of the respective pool of 24 patients. This carrier analysis led to the confirmation of 75 (65%) mutations/variants (‘true positives’), whereas 40 variants could not be confirmed (‘false positives’) (table 2). The distribution of ‘false’ versus ‘true positive’ were plotted in respect to the variant allele frequency (x axis) and the absolute sequence reads (counts) present for each of the 114 variants analysed (figure 3). The likelihood for a variant allele to be ‘a true positive’ was found to increase as expected, with higher absolute allele counts and/or higher relative variant allele frequency. In order not to miss mutations we chose relaxed SNP call parameters of 0.7% for the variant allele frequency with at least five absolute counts (reads). This has especially increased ‘false positive’ variants with allele frequencies between 0.7–1% (figure 3). CEL I heteroduplex analysis helped to identify 44 out of altogether 74 variants during the initial screen, but failed in 14 cases to identify the change, as subsequently revealed by Sanger sequencing (in tables 3 and 4 indicated as ‘CEL I negative’). All other variants/mutations have been identified solely by Sanger sequencing without CEL I prescreening. For patients in whom initially only one mutation has been identified, we searched for a second mutated allele by direct Sanger sequencing all exons of the respective gene. This approach led to the identification of 10 additional mutations.

Sanger sequencing confirmation of ‘true mutations/variants’ (filled diamonds) identified by MPR on Solexa/Illumina platform are plotted versus ‘false positive’ variants not confirmed (open circles). The alignment of a total of about 83 million sequence reads (39 bp each) to the human reference sequence of targeted 376 exons using ‘CLC Genomics Workbench’ software revealed 114 variants/mutations fulfilling the following criteria: (1) absent from single nucleotide polymorphism (SNP) database SNPv130; (2) absent from a control pool of 96 DNAs of healthy individuals; and (3) damaging impact at protein level predicted by PolyPhen2 with scores above 0.7. The variant frequency (x axis) is plotted against the variant counts (y axis) for each of the 114 variants analysed. Seventy-four variants/mutations have been confirmed by Sanger sequencing (filled diamonds) and we identified the respective mutation carrier out of a pool of 24 patients. Note that Sanger sequencing confirmation (filled diamonds) has been successful for only three variants with allele frequencies below 1%. The expected frequency of a heterozygous change found in a pool of 24 patients DNA is about 2.1%. Changes with frequencies above 1% and high absolute counts (>50) are almost always confirmed by Sanger sequencing.
Genotypes and phenotypes of 38 patients (30 families) with mutations in NPHP4, IQCB1, CEP290, RPGRIP1L, TMEM67, AHI1, CC2D2A, and TTC21B and respective MPR statistics
Genotypes and phenotypes of 24 patients carry a single heterozygous variant/mutation in at least one of 11 nephronophthisis associated ciliopathies (NPHP-AC) genes
Detection of disease causing mutations in 30 out of 120 patients
MPR mutation analysis of all coding exons of 18 different NPHP-AC genes (table 1) in 120 ascertained patients with a severe recessive NPHP-AC disease led to the identification of 57 mutated alleles, comprising 43 different mutations in 30 unrelated patients (table 3). Twenty-four patients carried a compound heterozygous mutation. In three patients with a consanguineous background, a homozygous disease allele was identified in CEP290 (p.L972P and p.G1890X twice) (table 3). In three patients, only one mutated allele (IQCB1, p.R364X; CEP290, p.K484fsX8; and AHI1, p.R891X) has been identified so far, even though all exons of the respective mutated gene have been sequenced directly. Mutations were discovered in the genes NPHP4 (2 patients), IQCB1/NPHP5 (1 patient), CEP290/NPHP6 (9 patients), RPGRIP1L/NPHP8 (1 patient), TMEM67/NPHP11 (10 patients), AHI1 (1 patient), CC2D2A (3 patients), and TTC21B (3 patients) (table 3). The category of mutated alleles identified in the present study are as follows: 14 nonsense mutations, 11 small insertion/deletions leading to a frameshift, 1 inframe deletion, 3 splice site mutations, and 25 missense mutations (table 3). Twenty-seven of these mutations were novel findings in the genes NPHP4 (p.N102fsX76, p.W1023X, p.T1122P, p.R1135fsX10), IQCB1 (p.R364X), CEP290 (p.E46X, p.G397S, p.K484fsX8, p.L972P, p.E1728X, p.E1771X, p.L1815fsX4), RPGRIP1L (p.N241fsX25, p.H610P), TMEM67 (p.K329T, p.R463X, p.F637L, p.V673A, p.Y723C, p.T964I, c.2556+1G→A splice), AHI1 (p.R891X), and CC2D2A (p.E229del, p.L559P, p.W1182R, p.E1259fsX1, p.V1298D) (table 3, figure 4). All mutations were absent from 96 healthy control individuals. In all cases where DNA of relatives was available, recessive mutations segregated with the affected status and segregated from parents as expected. In 17 out of 30 families segregation analysis has been performed and paternal (p) or maternal (m) inheritance has been indicated for respective mutations accordingly in table 3. The heterozygous TMEM67 mutation p.C615R was found recurrently in five different families of German origin. Comparison of SNP genotype linkage data, generated by 250k (StyI) Affymetrix SNP analysis, is compatible with extensive haplotype sharing between respective patients and extends up to 4.2 Mb (204 SNP markers), indicating inheritance from an ancestral founder (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sequence chromatograms of 28 different novel mutations identified in the genes NPHP4 (4), IQCB1 (1), CEP290 (8), RPGRIP1L (2), TMEM67 (7), AHI1 (1), and CC2D2A (5) in individuals with an NPHP-AC. Gene name, patient identifier, nucleotide change, and inferred amino acid alteration are given above sequence traces. Wild type sequence chromatograms are shown below mutated sequences. All mutations were absent from at least 96 healthy control individuals. Note that no second mutation has been identified in patients A394, A2420, and F787 in whom we identified a heterozygous truncating mutation in the genes IQCB1 (p.R364X), CEP290 (p.K484fsX8), and AHI1 (p.R891X), respectively. All mutations were found in the heterozygous state with the exception of a homozygous CEP290 missense mutation (p.L972P) in patient F335.
Single heterozygous variants of unknown significance
Mutation analysis by MPR of 18 known disease genes revealed an additional 26 single heterozygous missense mutations/variants of unknown significance in 24 patients out of the 120 patients analysed (table 4). All changes were absent from at least 96 healthy control individuals. For all 26 missense changes Sanger sequencing was performed for all exons of the respective disease gene, but failed to detect a second mutated allele. Patients of families A75 and A1421 carried heterozygous missense mutations/variants in different disease genes. In a patient from Poland (A75) with nephronophthisis, a heterozygous p.N85T mutation/variant in CEP290 together with a p.Y655C change in the gene RPGRIP1L was detected. The positions of both missense changes were evolutionarily conserved in vertebrates including zebrafish. PolyPhen2 software predicted a probable protein damaging effect for both missense changes (table 4). Individual A1421 from Egypt with JBTS and liver fibrosis carries the heterozygous missense changes in TMEM67 (p.G821S) and CC2D2A (p.R1019G). Interestingly, the mutation p.G821S has been found homozygously in patients with nephronophthisis with liver fibrosis, published recently (Otto et al, 2009)14. The amino acids of both missense changes found in family A1421 are highly conserved in evolution including the nematode Caenorhabditis elegans. Altogether, we identified 21 different single heterozygous mutations/variants in the genes NPHP3 (1), NPHP4 (2), IQCB1 (1), CEP290 (1), RPGRIP1L (2), TMEM67 (5), ARL13B (1), CC2D2A (3), TTC21B (3), MKS1 (1), and XPNPEP3 (1). The mutation p.G821S in TMEM67 and heterozygous changes p.K507E and p.P721S in the gene CC2D2A have been found recurrently (table 4). Five out of the 21 different single heterozygous changes have been published previously as disease causing in a recessive setting and are documented in the Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff (HGMD ‘Biobase’ (http://www.biobase-international.com/) (table 4).
Discussion
Applying next generation sequencing technology helped us to identify the disease causing mutations in 30 out of 120 patients with severe NPHP-AC. Altogether, we identified 43 different mutations, 28 of which were novel findings within nine different NPHP-AC genes. Additionally, we found single heterozygous missense changes of unknown significance in 25 patients in 11 out of 18 genes analysed. We demonstrated that high throughput sequencing using pooled DNA samples is highly efficient in detecting rare mutations in large cohorts of patients with diseases of broad genetic locus heterogeneity. Applying MPR, we screened 376 coding exons derived from 18 different NPHP-AC genes in 120 patients, which comprises more than 45 000 different amplified DNA fragments. Alternative methods to screen such a large amount of single PCR products, like standard Sanger sequencing, although accurate and reliable, are prohibitively expensive (US$180 000, £114 000, €130 000) and would require 45 000 single PCR amplifications. CEL1 heteroduplex screening alone without high throughput sequencing on the other hand is less expensive (estimated costs $12 000, £7600, €9000); however, it would require laborious and time consuming analyses of about 450 different 96-well plates. Another alternative approach is exon capture in combination with next generation sequencing, which might be considered when a large number of exons (or the total exome) have to be analysed for a reasonable number of DNA samples (about $4000(£2500, €3000)/exome/sample). In comparison, the cost of generating sequence data by MPR of five pooled samples (376 exons, 120 patients) and subsequent confirmation analyses in the present study was in the range of $6000 (£3800, €4400). The disadvantage of MPR is the high inherent error rate of about 0.5% per base call. This is problematic especially when samples are pooled and the expected variant frequencies are very low, which certainly will result in high false positive rates. Out of 114 mutations/variants which were predicted after alignment and next generation sequencing software analysis, only 74 (65%) have been confirmed by Sanger sequencing.
Recently, we applied the pooling and MPR strategy also to a cohort of 105 patients with Bardet–Biedl syndrome and screened for mutations in 12 BBS genes. Software and hardware improvements of the Illumina sequencers are constantly increasing the number and quality of sequence reads. Interestingly, 100% of the exonic bases in all BBS genes were covered sufficiently (>300x) with an average coverage of 19 711 after analysing the most recent sequence run with 28 million short reads. After alignment 11 out of 12 (92%) of the predicted mutations have been directly confirmed by Sanger sequencing (manuscript in preparation). The increased number of sequence reads and the resulting increased base coverage seems to reduce false positive calls dramatically. Better coverage can be also obtained by generating longer (eg, 78 bases) and/or paired-end sequence reads. This will further reduce calling false positives and will strongly reduce the number of confirmation experiments using CEL I endonuclease digestion.
In order to reliably detect a heterozygous variant/mutation in a pool of 48 alleles (expected frequency 2.1%) after generating a total of up to 800 Mb worth of sequence, we choose a cutoffs for minor allele frequency of 0.7% and a minimum count of five reads. In a pilot project with 18 pooled DNA samples from patients with already known mutations we were able to identify all but two out of 24 known mutated alleles using the outlined cut-off parameters. In both of these missed alleles, the coverage depth was insufficient due to PCR amplification problems.
The question arises as to how many mutations might have been missed in the experimental pooling and MPR approach in the 120 patients investigated. We have recognised insufficient coverage below 300× for about 5% of all coding nucleotides out of a total of the 52.8 kb sequenced and expect therefore at least 5% of mutations to be missed. MPR of PCR amplicons generated from pooled DNA samples revealed initially 114 potential mutations using CLC genomics workbench software for alignment and analysis. Seventy-four (65%) of these variants have been finally confirmed by Sanger sequencing, 41 of which represented single heterozygous changes only, without a second mutated allele identified initially. However, Sanger sequencing of all exons of the respective gene in carriers of these single heterozygous changes revealed the second recessive mutation in 10 patients. This indicates that we have missed at least 12% (10/87) of mutations/variants during the initial analysis. The calculated likelihood to miss both mutations of a patient with a compound heterozygous mutation is only in the range of 1–2% (0.12×0.12).
Besides the mutations we missed because of low coverage depth, there were three different 4 bp deletions with sufficient coverage which were missed using the CLC next generation sequence software. To address the problem of detection of small indels (four and more bases) in short reads (39 bp), the use of other software packages have to be considered like ‘NextGENe’ from the company Softgenetics or the program ‘Novoalign’ from Novocraft. For longer reads of, for example, 78 bp, we found that CLC next generation sequence software reliably calls indels of up to six bases. To date, the mutation rate of indels (four or more bases) in NPHP related genes published in the HGMD ‘Biobase’ mutation database (release 25 June 2010) is about 6%. Out of 457 different mutations published in the 18 NPHP related genes, 28 (6.1%) fall into this category of four or more bases inserted or deleted.
MPR revealed 24 patients with only one heterozygous missense change in one gene or two missense changes in two different genes (three families, table 4). We speculate that in most of these cases a second mutated allele has been missed, although all exons have been analysed by Sanger sequencing. Examples could be gross rearrangements, copy number variations, deep intronic splice affecting changes, promotor mutations, or polyadenylation signal variants, which were not detectable by coding exon sequencing alone. Some of the changes found might represent only rare polymorphism without any disease relevance. We have not found any evidence that oligogenicity is involved in NPHP-AC, although we sequenced 120 patients for all known relevant 18 NPHP-AC genes.
The approach of MPR of pooled samples presented here is robust, cost efficient, and best suited for screening large cohorts for mutations in genetically heterogeneous diseases. The lack of sensitivity seen so far for the MPR of pooled sample approach makes a clinical diagnostic application impracticable. In cases where numerous changes are expected or in a clinical mutation diagnostic setting, ‘barcoding tags’ offer an alternative and should be considered. However, that approach requires additional library preparations, large amounts of single PCR reactions or establishment of very complex multiplex PCRs reactions.40 Anticipated future sequencing technology improvements will allow parallel mutation analysis of higher numbers of genes and DNA samples. This is especially of interest because the lack of mutations in 75% of patients in our cohort indicates further extensive heterogeneity in NPHP-AC.
Acknowledgments
The authors sincerely thank the affected individuals and their families for participation and we thank the physicians who contributed to this study. We acknowledge RH Lyons for excellent next generation sequencing. FH is an Investigator of the Howard Hughes Medical Institute, a Doris Duke Distinguished Clinical Scientist, and a Frederick G. L. Huetwell Professor. This research was supported by grants from the National Institutes of Health to FH (DK1069274, DK1068306, DK064614). We thank all the physicians and researchers of the ‘Gesellschaft für Pädiatrische Nephrologie (GPN)’ study group for participation.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Contributing members of the GPN study group are: C Bergmann (Aachen, Germany); K Zerres (Aachen, Germany); J Gellermann (Berlin, Germany); A Münch (Berlin, Germany); L Neumann (Berlin, Germany); MJ Schürmann (Berlin, Germany); I Franke (Bonn, Germany); B Beck (Cologne, Germany); K Josefiak (Cologne, Germany); D Michalk (Cologne, Germany); Dr Stapenhorst (Cologne, Germany); T Ronda (Cologne, Germany); M Weber (Cologne, Germany); T Erler (Cottbus, Germany); B Weidner (Cottbus, Germany); KE Bonzel (Essen, Germany); A-M Wingen (Essen, Germany); J Dippell (Frankfurt, Germany); J Kirschner (Freiburg, Germany); R Korinthenberg (Freiburg, Germany); M Mall (Freiburg, Germany); H Omran (Freiburg, Germany); G Wolff, (Freiburg, Germany); S Fuchs (Hamburg, Germany); A Gal (Hamburg, Germany); M van Husen (Hamburg, Germany); S Lüttgen (Hamburg, Germany); DE Müller-Wiefel (Hamburg, Germany); J Drube (Hannover, Germany); JHH Ehrich (Hannover, Germany); S Fründ (Hannover, Germany); J Strehlau (Hannover, Germany); GF Hoffmann (Heidelberg, Germany); D Kiepe (Heidelberg, Germany); C Kneppo (Heidelberg, Germany); S Rieger (Heidelberg, Germany); B Tönshoff (Heidelberg, Germany); R Bambauer (Homburg, Germany); R Klüte (Ibbenbüren, Germany); M Heckel (Kronach, Germany); A Greiner (Leipzig, Germany); N Jeck (Marburg, Germany); R Roos (München, Germany); M, Bulla (Münster, Germany); S Fründ (Münster, Germany), B Frye (Münster, Germany); E Harms (Münster, Germany); E Kuwertz-Broeking (Münster, Germany); B Wittwer (Münster, Germany); R Sanwald (Pforzheim, Germany); H-J Stolpe (Rostock, Germany); J Höpfner (Schweinfurt, Germany); M Holder (Stuttgart, Germany); H-E Leichter (Stuttgart, Germany); G Baynam (Subiaco, Australia); C Edwards (Subiaco, Australia); H Peters (Victoria, Australia); C Jones (Victoria, Australia); A Janecke (Innsbruck, Austria); G Sunder-Plassmann (Vienna, Austria); K Devriendt, Leuven, Belgium); J Chow (Vancouver, Canada); P Trnka (Vancouver, Canada); K Õunap (Tartu, Estonia); T Apostolou (Athene, Greece); B Afroze (Kuala Lumpur, Malaysia); N Lock Hock (Kuala Lumpur, Malaysia); M Eccles (Otago, New Zealand); JW Dixon (Wellington, New Zealand; S Hashmi (Karachi, Pakistan); D Drozdz (Kraków, Poland); A Pogan (Kraków, Poland); A Peco-Antic (Belgrade, Serbia); B Milosevic (Novi Sad, Serbia); V Stojanovic (Novi Sad, Serbia); E Holmberg (Umea, Sweden); I Kern (Geneva, Switzerland); PH Axwijk (Amsterdam, The Netherlands); N Knoers (Nijmegen, The Netherlands); F Ozaltin (Ankara, Turkey); N Besbas (Ankara, Turkey); M Koyun (Antalya, Turkey); A Nayir (Istanbul, Turkey); H Kayserili (Istanbul, Turkey); S Ozturk (Istanbul, Turkey); D Pehlivan (Istanbul, Turkey); R Farrington (Cambridge, UK); FL Raymond (Cambridge, UK); R Sandford (Cambridge, UK); J Whittaker (Cambridge, UK); B Kerr (Manchester, UK); M Cadnapaphornchai (Denver, CO, USA); G Hidalgo (Detroit, MI, USA); S Andreoli (Indiananapolis, IN, USA); B Mills (Indiananapolis, IN, USA); M Bendel-Stenzel (Minneapolis, MN, USA); N Stover (Portland, OR, USA); R Weleber (Portland, OR, USA); M DeBeukelaer (Toledo, OH, USA); C Kozma (Washington, DC, USA); R Schonberg (Washington, DC, USA); M Bitzan (Winston-Salem, NC, USA).
Funding Other funders: NIH; Howard Hughes Medical Institute.
Competing interests None to declare.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the University of Michigan.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Correction