Article Text

Abstract
Background Mutations in SOD1, ANG, VAPB, TARDBP and FUS genes have been identified in amyotrophic lateral sclerosis (ALS).
Methods The relative contributions of the different mutations to ALS were estimated by systematically screening a cohort of 162 families enrolled in France and 500 controls (1000 chromosomes) using molecular analysis techniques and performing phenotype–genotype correlations.
Results 31 pathogenic missense mutations were found in 36 patients (20 SOD1, 1 ANG, 1 VAPB, 7 TARDBP and 7 FUS). Surprisingly two FUS mutation carriers also harboured ANG variants. One family of Japanese origin with the P56S VAPB mutation was identified. Seven novel mutations (three in SOD1, two in TARDBP, two in FUS) were found. None of them was detected in controls. Segregation of detected mutations with the disease was confirmed in 11 families including five pedigrees carrying the novel mutations. Clinical comparison of SOD1, TARDBP, FUS and other familial ALS patients (with no mutation in the screened genes) revealed differences in site of onset (predominantly lower limbs for SOD1 and upper limbs for TARDBP mutations), age of onset (younger with FUS mutations), and in lifespan (shorter for FUS carriers). One third of SOD1 patients survived more than 7 years: these patients had earlier disease onset than those presenting with a more typical course. Differences were also observed among FUS mutations, with the R521H FUS mutation being associated with longer disease duration.
Conclusions This study identifies new genetic associations with ALS and provides phenotype–genotype correlations with both previously reported and novel mutations.
- ALS
- family study
- genetics
- phenotype
- mutation
- clinical genetics
- molecular genetics
- motor neuron disease
- neuromuscular disease
Statistics from Altmetric.com
- ALS
- family study
- genetics
- phenotype
- mutation
- clinical genetics
- molecular genetics
- motor neuron disease
- neuromuscular disease
Introduction
Recent genetic advances have brought to five the number of genes involved in typical amyotrophic lateral sclerosis (ALS). The first identified was the SOD1 gene, which encodes cooper/zinc superoxide dismutase.1 More than 130 mutations, which account for 12–23% of familial ALS (FALS), have now been reported (http://alsod.iop.kcl.ac.uk/Als/Index.aspx). A single mutation, c.166C>T/Pro56Ser (P56S), in the gene encoding VAMP (vesicle associated membrane protein) associated protein B (VAPB), was subsequently identified in Brazilian kindreds who shared a common founder from the time of the Portuguese colonisation of Brazil.2 3 The recent discovery of mutations in the ANG (angiogenin),4 TARDBP (TAR DNA binding protein 43)5–11 and FUS (fused in sarcoma)12 13 genes, all encoding proteins probably involved in RNA metabolism, have opened new areas of study into possible molecular mechanisms underlying ALS.
In the present study, we establish the frequencies of mutations in these five genes by systematically screening a cohort of 162 French patients with FALS, and compare the phenotypes related to the different mutations.
Methods
Patients
The population comprised 162 index cases of unrelated families with probable or definite ALS,14 mainly of Caucasian origin (except three patients of Asian and one Sepharadic Jewish heritage). DNA samples were collected over the past 15 years at the ALS National Reference Center of Pitié-Salpêtrière Hospital (Paris). During the same period, the number of ALS patients followed up by the centre was 6602. Relatives of some families for which we identified a mutation were collected by other French ALS centres belonging to the French ALS study group. One hundred patients were male and 62 were female (M:F ratio=1.61:1). Control samples were obtained from 500 healthy Caucasian individuals matched to the sex and age of the patients. Protocols were approved by the ‘Comité d'Ethique de la Pitié-Salpêtrière’ and by the Medical Research Ethics Committee of ‘Assistance Publique-Hôpitaux de Paris’. All participants signed a consent form for the research.
Molecular analysis
Coding regions and exon–intron boundaries of SOD1 (5 exons), ANG (exon 1 and 2), VAPB (5 exons), TARDBP (5 exons), and FUS (15 exons) genes were amplified from genomic DNA in 5, 3, 5, 6 and 13 fragments, respectively. Sequence of the primers and amplification conditions are available upon request.
Clinical features
The following clinical data were collected: age at onset (first symptom of weakness), site of onset (upper limb, lower limb, bulbar), presence of cognitive impairment15 and survival. For survival we used two variables bringing complementary information for mutation carriers: the disease duration (interval from onset to death or censoring) and lifespan (age at death or censoring), defined by the delay between birth and death. This parameter appeared informative in FALS patients as it could provide an insight into the duration of the whole disease process. Features recorded at the first clinical evaluation (initial signs) and at the last evaluation (final signs) were: predominance of lower motor neuron (LMN) or upper motor neuron (UMN) involvement (according to El Escorial criteria)14 and bulbar function (using the modified Norris bulbar scale).16 To assess the degree of deterioration in bulbar function during the course of the disease, we calculated a bulbar score (difference between initial and final Norris bulbar scale scores).
Statistical analysis
Mean and median age of onset, disease duration and lifespan were determined using maximum likelihood estimates including censored data. Data were censored at the last date of analysis (1 February 2010) if the patient was alive. The Cox proportional hazards regression model examined the relationship between potential predictors and the outcome of interest (either age at onset, disease duration or lifespan). Mutation status (SOD1, TARDBP, FUS, other FALS) was treated as a categorised variable. If the difference was significant (p<0.05), a log rank analysis compared groups by pairs. A similar analysis compared disease duration in FUS patients carrying either the R521H or other mutations. Proportions of patients classified according to gender, onset site, disease duration and presence/absence of cognitive impairment were compared by pairs between SOD1, TARDBP, FUS and other FALS groups using Fisher's exact tests. A Mann–Whitney U test compared mean age at onset between two subgroups of SOD1 patients (with disease duration >7 or ≤7 years) and mean disease duration in patients carrying the R521H or other FUS mutation. One way analysis of variance (ANOVA) tests compared mean bulbar scores in SOD1, TARDBP and FUS patients. If the difference was significant (p<0.05), pairwise comparisons were made using a Tukey–Kramer post test. Statistical analyses were performed using the SPSS 11.0 data analysis software (SPSS Inc).
Results
Genetic data
We sequenced the SOD1, ANG, VAPB, TARDBP and FUS genes simultaneously in all patients.
We found 18 different SOD1 missense mutations in 20 out of the 162 FALS patients, corresponding to a mutation frequency of 12.3% (supplementary table 1). Table 1 summarises the mutations we found and indicates the correspondence between the numbering of SOD1 mutations according to Human Genome Variation Society guidelines (http://www.hgvs.org/) and the ALS Online Database (http://alsod.iop.kcl.ac.uk/Als/index.aspx). Three patients harboured novel SOD1 mutations, that were not detected in controls and that affected a highly conserved residue until xenopus: c.139C→G/p.His47Asp (H46D), c.200C→G/p.Pro67Arg (P66R) and c.251A→G/p.Asp84Gly (D83G). The affected relatives were deceased so the segregation of H46D and P66R mutations could not be established. The D83G mutation was present in an affected brother of the index case, but not in the healthy sister (supplementary figure 1A).
The 31 identified mutations in the 162 familial amyotrophic lateral sclerosis (FALS) index cases
Next, we identified the same c.122A→T/pLys41Ile (K17I) ANG mutation in two families with dominant inheritance. The K17I mutation has been reported in three apparently healthy people,4 17 but was not detected in our 500 controls. We also found the c.232A→G/p.Lys78Glu (K54E) and the c.434G→A/p.Arg145His (R121H) variants in two FALS, which were previously reported as mutations in sporadic ALS (SALS).17 18 In the present study, however, these substitutions were not detected in the other affected relatives. One of the K17I carriers was later found to carry a R521C FUS mutation and the K54E carrier was found to carry a new FUS mutation (R521S), which segregated with the disease (supplementary figure 1H).
We also identified the c.166C→T/p.Pro56Ser (P56S) VAPB mutation in one family, giving a mutation frequency of 0.6%. The P56S carrier was of Japanese descent, and represents the first non-Brazilian patient carrying this mutation. Three other members of his family had motor neuron disease, suggesting autosomal dominant transmission. We did not detect any other variant of this gene in FALS. The finding of a P56S VAPB mutation in a patient of Japanese origin presumably reflects the Portuguese trading connection with the Far East and Brazil in the mid 16th century.
Six different TARDBP missense mutations, all located in exon 6, were detected in seven families (table 1), corresponding to a mutation frequency of 4.3%. Pedigrees of the G295S, the A382T and the A315T substitutions have been described elsewhere.6 19 An additional family carrying the G348C mutation was identified. This mutation was detected in the mother of the index case, an obligate carrier who has not developed the disease at age 65 (supplementary figure 1G). We also identified two novel mutations: the c.1150G→C/p.Gly384Arg (G384R) mutation in an affected woman, her affected sister, and in an obligate carrier aged 73 years who has not developed the disease but who has transmitted it to her affected son (supplementary figure 1E); and the c.1153T→G/p.Trp385Gly (W385G) mutation in two affected brothers of a separate family (supplementary figure 1F). These two novel mutations seemed to be pathogenic as they co-segregated with the disease, they were not detected in 500 controls, and the amino acids targeted, Gly384 and Trp385, were conserved until xenopus.
We identified five different missense mutations in FUS in seven families. Two of these mutations were novel (table 1). The frequency of FUS mutations was 4.3% of our FALS patients. All FUS mutations were located in exon 15. No mutation was found in controls. Four mutations (R521C, R521H, R521S, R521L) targeted the same arginine residue that is conserved until xenopus. The first novel FUS mutation was a c.1561C→A/p.Arg521Ser (R521S) substitution. The mutation was absent in the healthy sister of the index case but present in a far cousin who died from a bulbar ALS (supplementary figure 1H). The second novel mutation was a c.1562G→T/p.Arg521Leu (R521L) mutation. DNA samples of three healthy relatives (father and two aunts) did not bear the mutation, confirming that the disease was transmitted to the index case by her affected mother (supplementary figure 1I). In both families, the penetrance was incomplete as the disease was transmitted by obligate carriers who lived until the age of 73 and 77 without developing ALS. The already reported c.1561C→T/p.Arg521Cys (R521C), c.1562G→A/p.Arg521His (R521H) and c.1542G→T/p.Arg514Ser (R514S) mutations were also detected. We confirmed the segregation of the R521H mutation (supplementary figure 1J) and showed, for the first time, the segregation of the R514S mutation (supplementary figure 1K). Surprisingly an FUS mutation was identified in two patients who also carried K17I or K54E ANG variant (table 1).
Clinical findings
Clinical data could be recovered for 100 relatives belonging to the 36 families with mutations and is summarised for 81 patients, from whom the information was the most complete, in supplementary table 2.
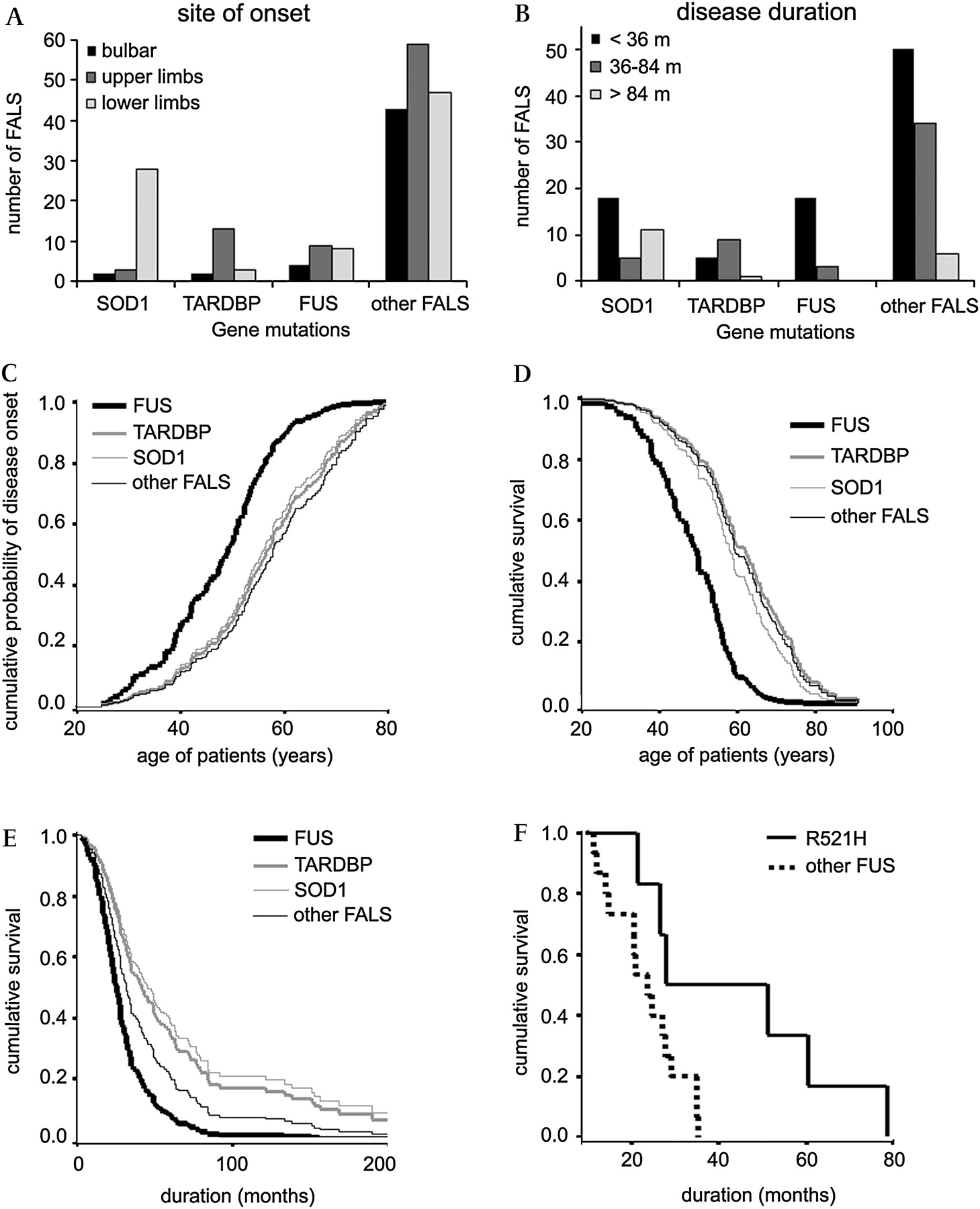
Age at onset for the SOD1, TARDBP, FUS and other FALS (with no mutation in the screened genes) groups was statistically different (table 2, figure 1C). FUS patients had younger onset than SOD1 (log rank: p=0.0111) and other FALS (log rank: p<0.0001) patients.
Disease characteristics by mutated gene
{kind=link}
Distribution of each group of patients by age of onset, site of onset, disease duration and lifespan. (A) Histograms showing the distribution of patients with SOD1, TARDBP or FUS mutations and other familial amyotrophic lateral sclerosis (FALS) patients according to bulbar (black), upper limb (dark grey) or lower limb (light grey) onset. (B) Histograms illustrating the distribution of SOD1, TARDBP, FUS or other FALS patients by disease duration (in months): black: <36 months, dark grey: 36–84 months, light grey: >84 months. Cox regression curves of (C) cumulative probability of disease onset according to the age of patients, (D) cumulative survival probability according to the age of patients, and (E) cumulative survival probability from time of disease onset. (C–E) The four patient groups were SOD1 (grey thin line), TARDBP (grey bold line), FUS (black bold line), and other FALS (black thin line). The graph was truncated at 200 months in (E) to improve visualisation at short time points. (F) Kaplan–Meier plots of cumulative survival from time of disease onset of patients carrying different FUS mutations: either R521H (bold line) or other mutations combined (dotted line).
Lifespan for the SOD1, TARDBP, FUS and other FALS patient groups was significantly different (table 2, figure 1D). FUS patients had a shorter lifespan than SOD1 (log rank: p=0.0002), TARDBP (log rank: p=0.0028) or other FALS (log rank: p<0.0001) patients. The SOD1 and TARDBP groups were similar.
Disease duration of the SOD1, TARDBP, FUS and other FALS groups was different (table 2, figure 1E). The disease was more rapid in FUS than in SOD1 (log rank: p=0.0082), TARDBP (log rank: p=0.0019) or other FALS patients (log rank: p=0.0425). To further examine disease duration, we separated the mutated patients into three groups: ‘rapid course’ (disease duration <3 years), ‘medium course’ (3–7 years), and ‘slow course’ (>7 years). Our SOD1 patients had heterogeneous disease durations with bimodal distribution; more than half (53%, n=18) had a rapid course as in some patients carrying the novel H46D and D83G mutations. One third of SOD1 patients (32%, n=11) had slow disease progression (figure 1B). The average age at onset was lower in this group (41.2±3.3, n=11) than in patients with more rapid course (56.4±2.3, n=23, Mann–Whitney U test: p=0.0017). Slow progression was notably observed in some patients with the novel P66R (>27 years) and D83G (>12 years) mutations and with the poorly documented N139D mutation. These patients typically had young onset and prominent LMN signs resembling progressive muscular atrophy. Long disease duration was also observed in the family with a VAPB mutation, who, as a whole, had leg onset in the sixth decade (supplementary table 2). Among the 15 patients with TARDBP mutations, only one presented with a slow course; two thirds (n=9) had a medium course and one third (n=5) developed rapid disease (figure 1B). No FUS patient had slow disease progression. More than 85% (n=18) died after a rapid disease (figure 1B). Three patients with R521H mutations had a ‘medium course’ leading to a disease duration longer for this FUS mutation than for the others (supplementary table 2, log rank: p=0.0213, figure 1F). The percentage of patients in each disease duration subgroup (<3, 3–7, >7 years) was compared between SOD1, TARDBP, FUS and other FALS patients using Fisher's exact test analyses. FUS patient distribution was different from that of SOD1 (Fisher's test: p=0.006), TARDBP (Fisher's test: p=0.002) and other FALS (Fisher's test: p=0.044). SOD1 patients distribution was also different from that of TARDBP (Fisher's test: p=0.0063) and other FALS (Fisher's test: p=0.0005).
The site of onset in FUS patients was in the arms (43%, n=9), legs (38%, n=8) or bulbar muscles (19%, n=4), which is similar to the other FALS groups. Site of onset (figure 1A) was mainly in the lower limbs for SOD1 (85%, n=28/33, Fischer's exact test for the difference with FALS group: p<0.0001) and in the upper limbs for TARDBP patients (76.4%, n=13/17, Fisher's exact test for the difference with FALS group: p=0.04). Initial and final predominance of UMN or LMN signs in the limbs was also evaluated (supplementary table 2). For SOD1 patients, LMN signs usually predominated. Most of the TARDBP patients initially presented with both UMN and LMN signs; with disease progression, LMN signs became predominant. One patient carrying the novel W385G mutation (exhibiting the longest survival time) developed final predominance of limb spasticity. FUS patients showed continuous predominance of LMN involvement except for the R521H patients with prolonged survival who developed pronounced UMN signs at onset. ANOVA tests used to compare mean bulbar scores between SOD1, TARDBP, FUS and other FALS groups showed a significant difference (table 2). Worsening of bulbar function with time tends to be less severe for the SOD1 and FUS groups compared to both TARDBP and other FALS patients (table 2).
No clinically apparent frontotemporal dysfunction occurred in SOD1 patients (table 2). Two out of 10 patients with TARDBP mutations demonstrated cognitive impairment that met criteria for frontotemporal dementia (FTD). One with the G295S mutation had FTD 2 years before developing bulbar motor signs.19 The other (with the novel G384R TARDBP mutation) developed FTD 1 year after the onset of motor weakness. A single patient carrying the R521H FUS mutation presented with cognitive impairment 5 months after the onset of ALS. The proportion of patients with cognitive impairment was not statistically different between the SOD1, TARDBP, FUS and other FALS patients (table 2).
Discussion
This study represents the first large scale investigation of FALS among the French population and the first report comparing the phenotypes of the recently identified ALS genes. A mutation was identified in 36 out of the 162 tested families (22.2%). The overall percentages of SOD1 (12.5%), ANG (0.6%), VAPB (0.6%), TARDBP (4.3%) and FUS (4.3%) mutations were relatively close to those previously reported in European populations (supplementary table 1).
All of the 31 different mutations reported here affected evolutionarily conserved residues in the corresponding proteins and were absent from 500 controls (1000 chromosomes). The segregation with the disease could be established in 11 families (four SOD1, three TARDBP and four FUS). The co-segregation of 11 TARDBP mutations5 6 8–10 19–23 and six FUS mutations12 13 24 25 with ALS has been previously demonstrated. In this report, we extend these lists to include two new TARDBP mutations (G384R and W385G), two new FUS mutations (R521S and R521L), as well as the R514S FUS mutation. The extension of our pedigrees should allow confirming the segregation of the disease with these mutations. Indeed labelling a variant as pathogenic is a danger. We found two index cases harbouring ANG variants (K54E and R121H) that did not segregate with disease and we concluded that they were not the cause of the disease in these FALS. Moreover we found a FUS mutation in two index cases harbouring ANG variants (K54E or K17I). These results suggest that, as for others,26 the association of some ANG variants and ALS is questionable.
Some of the mutations we identified demonstrated variable penetrance. For example, we confirmed the presence of TARDBP mutations in two obligate female carriers who transmitted the disease to their children but who were asymptomatic at age 65 and 73.
The number of patients allowed comparison of phenotypes in relation to the involved gene. Although these findings have to be confirmed in larger populations, we could find statistical differences for some clinical traits between the different mutation groups. Patients with SOD1 mutations usually first developed lower limb weakness whereas patients with TARDBP mutations had onset predominantly in the upper limbs. Patients harbouring FUS mutations had various onset sites in the arms, legs or bulbar muscles, which resembles typical ALS more.
Evolution of the disease was categorised as rapid (<3 years), medium (3–7 years), or long (>7 years) for each gene. FUS mutations seemed to lead to the most aggressive disease, with a young onset and a rapid course for most of the patients, an observation that is consistent with previous reports.12 13 Analyses also showed that lifespan is shortened for FUS patients. Among patients with FUS mutations, three with the R521H mutation had a disease course that was approximately twofold longer than the others (supplementary table 2). Our results also showed that bulbar deterioration and cognitive impairment tended to be less frequent in patients harbouring SOD1 and FUS mutations.
Considerable intrafamilial phenotypic differences were observed in some families carrying various mutations in the SOD1, TARDBP and FUS genes. Age and site of onset varied between members of a family. The disease duration could also differ, as shown in families carrying the D83G SOD1 mutation (6–151 months in two siblings), the W385G TARDBP mutation, and the R521H FUS mutation. These data support the hypothesis that a mutation is not the only factor that determines the clinical course of the disease. Other factors must also contribute to phenotype, and it is not yet possible to predict the evolution of patients based solely on presence of the mutation or rate of progression in other family members.
Altogether our results show that one determinant of ALS phenotype is the underlying causative mutation. However, heterogeneity between and among families implies that other environmental and genetic influences contribute to not only the rate of evolution and which signs predominate, but also whether the disease will appear at all during life. Despite recent advances in the field of ALS genetics, most of the genes involved in FALS are still unknown. Considerable work lies ahead in determining the genetic and environmental factors that most contribute to ALS.
Acknowledgments
We are grateful to the patients and their families. We thank Joelle Debusne, Eliane Gardais, Luzia Vacherie, Thierry Larmonier and Dr Safaa Saker (Genethon cell and DNA bank), and Christelle Dussert, Isabelle Lagroua, Sylvie Forlani and Dr Alexandra Dürr (CRICM DNA and cell bank) for patient DNA, Patricia Bouillon for searching archival documents, and Elodie Chabrol for kind technical help. This study was supported by the Association pour la Recherche sur la sclérose latérale amyotrophique et autres maladies du motoneurone (ARS).
References
Supplementary materials
Web Only Data jmg.2010.077180
Files in this Data Supplement:
Footnotes
Funding Other Funders: Association pour la Recherche sur la sclérose latérale amyotrophique et autres maladies du motoneurone (ARS).
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the “Comité d'Ethique de la Pitié-Salpêtrière” and the Medical Research Ethics Committee of “Assistance Publique-Hôpitaux de Paris”.
Provenance and peer review Not commissioned; externally peer reviewed.