Article Text

Abstract
Background Deletion and the reciprocal duplication in 16p11.2 were recently associated with autism and developmental delay.
Method We indentified 27 deletions and 18 duplications of 16p11.2 were identified in 0.6% of all samples submitted for clinical array-CGH (comparative genomic hybridisation) analysis. Detailed molecular and phenotypic characterisations were performed on 17 deletion subjects and ten subjects with the duplication.
Results The most common clinical manifestations in 17 deletion and 10 duplication subjects were speech/language delay and cognitive impairment. Other phenotypes in the deletion patients included motor delay (50%), seizures (∼40%), behavioural problems (∼40%), congenital anomalies (∼30%), and autism (∼20%). The phenotypes among duplication patients included motor delay (6/10), behavioural problems (especially attention deficit hyperactivity disorder (ADHD)) (6/10), congenital anomalies (5/10), and seizures (3/10). Patients with the 16p11.2 deletion had statistically significant macrocephaly (p<0.0017) and 6 of the 10 patients with the duplication had microcephaly. One subject with the deletion was asymptomatic and another with the duplication had a normal cognitive and behavioural phenotype. Genomic analyses revealed additional complexity to the 16p11.2 region with mechanistic implications. The chromosomal rearrangement was de novo in all but 2 of the 10 deletion cases in which parental studies were available. Additionally, 2 de novo cases were apparently mosaic for the deletion in the analysed blood sample. Three de novo and 2 inherited cases were observed in the 5 of 10 duplication patients where data were available.
Conclusions Recurrent reciprocal 16p11.2 deletion and duplication are characterised by a spectrum of primarily neurocognitive phenotypes that are subject to incomplete penetrance and variable expressivity. The autism and macrocephaly observed with deletion and ADHD and microcephaly seen in duplication patients support a diametric model of autism spectrum and psychotic spectrum behavioural phenotypes in genomic sister disorders.
- Developmental delay
- autism
- copy number variations
- macrocephaly and microcephaly
- epilepsy
Statistics from Altmetric.com
Introduction
Array comparative genomic hybridisation (aCGH) allows a comprehensive interrogation of thousands of discrete genomic loci for segmental genomic copy number variations (CNVs). The clinical applications of aCGH and single nucleotide polymorphism (SNP) arrays have revolutionised the diagnostic work-up of patients with global developmental delay (GDD), mental retardation (MR), autism (MIM 209850), multiple congenital anomalies (MCA), and dysmorphic features, and facilitate disease gene discovery and prenatal diagnostics.1 The detection rates of clinically significant de novo chromosomal imbalances among GDD/MR patients can be as high as 20%.2 Recent studies show that de novo deletions and duplications play a significant role in the aetiology of syndromic and idiopathic autism, with a detection yield ranging between 7–30%.3–5 An interesting fact that emerged from the aggregate studies of MR, GDD and autism is that many of the deletions or duplications were de novo with a low recurrence rate in siblings, emphasising the genetic and genomic heterogeneity of MR/GDD and autism and the need for whole genome methods to detect these genomic changes.
Deletion and duplication syndromes, also known as genomic disorders,6 7 represent recurrent chromosomal abnormalities that are associated with distinct phenotypic features. These deletions/duplications often occur between low copy repeats (LCRs) and are commonly caused by non-allelic homologous recombination (NAHR) events.8
‘Forward genomics’ enables the identification of a specific CNV locus associated with a clinical phenotype assayed in the genetic screen. ‘Reverse genomics’, comparisons of the observed clinical features in patients with a specific genomic interval that is rearranged,9 enables clinical characterisation of previously unknown genomic disorders. In such studies, the identification of similar genomic imbalances in many patients with a recognisable phenotype can help clarify the role of these genomic changes in causing the specific clinical features. The detection of a de novo genomic imbalance in a single patient is not sufficient to identify a syndrome or prove pathogenicity of a given CNV. The number of novel genomic disorders that are being discovered by aCGH is growing continuously and includes the 16p11.2–p12.2,10 15q24,11 17q21.31,12–15 and 15q13.316 deletion syndromes. In some of these syndromes, the clinical heterogeneity and incomplete penetrance are remarkable, as has been demonstrated regarding the 15q13.3 microdeletion syndrome17–21 and in patients with the 1q21.1microdeletions and microduplications.22 23 Interestingly, deletions of both 15q13.3 and 1q21.1 have recently been associated with schizophrenia.18 19
Recently, a de novo recurrent 593 kb deletion and a de novo or inherited reciprocal duplication on 16p11.2 were associated with autism and DD.5 24 25 These CNVs are located at a rearrangement hot spot caused by flanking 147 kb duplicated blocks of LCRs which confer genomic instability and can lead to unequal crossing over via NAHR during meiosis.8 25 Weiss et al, using three populations, reported 16p11.2 deletions and duplications in ∼1% of autism subjects and 1.5% of children diagnosed with developmental or language delays.25 Marshall et al observed a similar 1% frequency among patients with autism.5 However, these studies did not present a detailed phenotypic characterisation of individuals with the 16p11.2 imbalances. A more recent study showed that autism was not the presenting feature in many patients with the 16p11.2 deletions and that these deletions are associated with a variable clinical spectrum ranging from MR and/or MCA, autism, and learning and speech problems, to a normal phenotype.26
Patients and methods
Patients
Patients with 16p11.2 rearrangements were recruited to this study by their referring physicians. The Medical Genetics Laboratories (MGL) at Baylor College of Medicine (BCM) have performed aCGH on ∼7400 samples after probes specific for the 16p11.2 region were introduced to this clinical microarray. The samples were analysed on consecutive versions of targeted oligonucleotide based arrays, as described.27–29 A total of 27 deletion cases and 18 duplication cases were detected with a frequency of ∼0.6% (45/7400) in samples submitted to the diagnostic laboratory. Detailed clinical information and informed consent were available for 17 deletion patients and for 10 patients with duplications. Four of the deletion cases (individuals 1, 2, 4, 5) and one of the duplication subjects (10) were tested in other genetic laboratories and were part of our cohort. Experienced medical geneticists assessed the clinical phenotypes of the patients (11 patients were clinically evaluated by the same clinician, MS). Studies were approved by the Institutional Review Board (IRB) of Baylor College of Medicine and all participants or parental guardians were recruited after informed consent. Photographs of the patients were collected after obtaining informed consent for publication of the photographs in the medical literature.
Targeted clinical aCGH and custom high resolution 16p oligonucleotide array
Clinical samples were analysed on a series of progressively more complex arrays from December 2007 to March 2009. Initial arrays (CMA v.6.4 and up) were 44K oligonucleotide based (Agilent Technologies, Santa Clara, California, USA) partly in a bacterial artificial chromosome (BAC) emulation format as described previously27–29; more recently a 105K custom Agilent array has been used. Initial identification of the cases by aCGH was based on loss or gain of a genomic copy number interrogated by BAC clone RP11-301D18 or by oligonucleotides emulated to this BAC clone (chr16: 29,683,644-29,869,247).
We also designed an Agilent customised HD-CGH microarray in 8X15K format (G4427A) to specifically interrogate 16p11.2. Probes (9517) were selected from Agilent's HD CGH probe database to cover the short arm of chromosome 16, with a biased high density over the region chr16: 28 800 000–30 500 000 (average spacing 0.65 kb compared with 5.3 kb for the rest of the short arm of chromosome 16). All of the probes were melting temperature (TM) matched and were selected with the most stringent similarity filter, which filters out all probes with secondary genomic alignments in the NCBI36 build of the human genome sequence. In addition, we selected an additional 4745 probes from chromosomes 1–22 as internal controls.
Labelling, hybridisation, and analysis of microarrays
Probe labelling and hybridisation were performed following the manufacturer's protocol (Agilent Oligonucleotide Array based CGH for Genomic DNA Analysis, version 4.0 plus, the 8X15K complementary protocol unique to the eight-pack format) with modifications. Genomic DNA was isolated from peripheral blood with a Puregene kit (Gentra Systems, Minneapolis, MN). aCGH analyses were performed according to the manufacturer's protocol (Agilent Technologies). Briefly, 2 μg (clinical array) or 0.9 μg (custom 16p microarray) of genomic patient and reference DNA were digested with AluI (5 U) and RsaI (5 U) (Promega, R6281 and R6371) for 2 h at 37°C. Digestions were verified by agarose gel electrophoresis. Labelling reactions were performed according to the manufacturer's instructions (BioPrime Array CGH Genomic Labelling Module, Invitrogen Corporation, 18095-012; Perkin-Elmer Las Inc. Cyanine Smart Pack dCTP, PerkinElmer, NEL620001KT) with Cy5-dCTP for patient DNA and Cy3-dCTP for reference DNA. Each dye labelled DNA was purified using Microcon YM-30 filters (Millipore, Billerica, Massachusetts, 42410). Labelling efficiency was determined using a NanoDrop ND-1000 UV-VIS spectrophotometer. Each dye labelled patient and reference DNA was combined with 2 μg of human Cot-1 DNA (Invitrogen, Carlsbad, California, 15279–011), Agilent Blocking Agent (5190-0405), and Agilent hybridisation buffer (5188-6420). These mixtures were denatured at 95°C for 3 min, pre-incubated at 37°C for 30 min, and hybridised to the array in a hybridisation chamber (Agilent, G2534A) for 40 h at 65°C in a rotating oven (Agilent, G2545A). Arrays were washed using Agilent Oligo CGH Wash Buffer 1 and 2 (5188–5226), Acetonitrile (Sigma-Aldrich, 271004-1L), and Stabilisation and Drying Solution (Agilent, 5185–5979), according to the Wash Procedure B in Agilent's protocol. Slides were scanned on a GenePix 4000B Microarray Scanner (Axon Instruments, Sunnyvale, CA). Data were extracted from the microarray image, the background subtracted, and then normalised using Agilent Feature Extraction Software 9.5.3.1. These data were subsequently imported into array CGH Analytics Software v4.0.85 (Agilent Technologies). The genomic copy number was defined by the analysis of the normalised log2 (Cy5/Cy3) ratio average of the CGH signal. The moving average was computed using a 10 kb window. Regions that reached a threshold >0.4 were interpreted as a duplication, whereas thresholds less than −0.8/−3 were interpreted as heterozygous/homozygous deletion. Genomic region analyses were performed according to the human reference sequence build 36.1 (UCSC genome browser: http://genome.ucsc.edu) with the software provided by Agilent.
Fluorescence in situ hybridisation analysis
The 16p11.2 rearrangements detected by clinical aCGH were confirmed in all cases by fluorescence in situ hybridisation (FISH). Peripheral blood samples from the patients and their parents were obtained and whole blood lymphocytes were cultured after phytohaemagglutinin (PHA) stimulation using standard methods.
16p11.2 LCR structure analysis
Using the Segmental Duplication track of the http://genome.ucsc.edu browser (Human Genome Build 36.1), we performed an analysis of duplicated genomic sequences including known LCRs (segmental duplication >1 kb of non-repeat masked sequence with over 90% similarity), comparing the ∼1.3 Mb region surrounding the proximal 16p11.2 locus (chr16: 29 100 000–30 400 000) against itself.
Head circumference analysis
Head circumferences (HC) were measured as the maximal occipito-frontal head circumference using standard techniques. Age and sex matched HC, weight, and height (Hgt) centiles were obtained using Abase, a PalmOS based calculator, and converted into Z scores.30 Absolute macrocephaly is defined as HC ≥1.88 SD above the mean for age and sex.31 Relative macrocephaly is calculated by adjusting the HC to the Hgt (zHC is two or more SDs greater than standardised height).31 Microcephaly is defined as HC ≤1.88 SD below the mean for age and sex.31 The two groups (deletion cases, duplication cases) were compared to a Z score of zero (the population mean) using a one sample t test, and against each other using an unpaired two tailed t test with Welch's correction for unequal variances. Analysis and graphing was carried out in GraphPad Prism 5. We used the Z test for two proportions to compare the frequency of the different phenotypes among patients with deletions and duplications.
Results
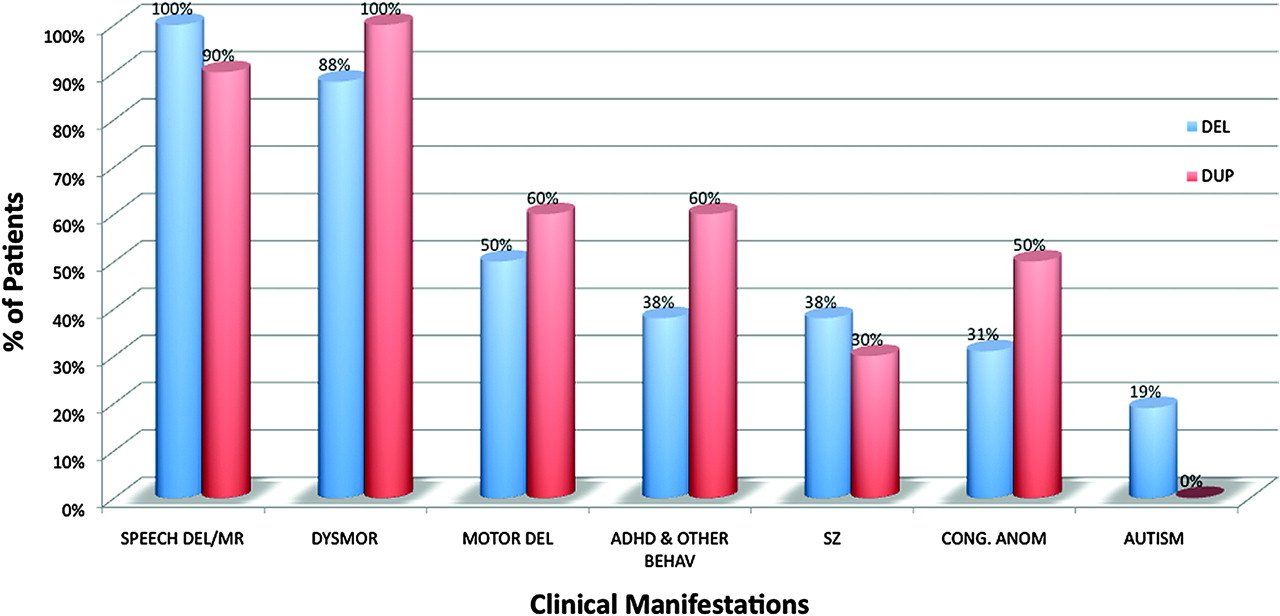
Twenty-seven index cases with 16p11.2 deletion and 18 index cases with 16p11.2 duplication were detected using aCGH during a period of 16 months in a total of 7400 samples referred to the diagnostic laboratory at Baylor College of Medicine. Each case was determined by aCGH to have a loss (deletion) or gain (duplication) corresponding to chromosomal coordinates (chr16:29 528 190–30 107 184). All aCGH clinical studies were confirmed by FISH analysis using the BAC clone RP11-301D18 within the proximal 16p11.2 region. The most common indications for testing were developmental delay/MR, dysmorphic features, seizures, congenital anomalies, autism, attention deficit hyperactivity disorder (MIM 143465) (ADHD)/other behavioural problems, and failure to thrive (figure 1). We found that dysmorphism (Z value 1.9, confidence level 97.1%) and ADHD/other behavioural problems (Z value 2.36, confidence level 99.1%) were statistically more common indications for testing of patients with the 16p11.2 duplications as compared to deletions. Although seizures and congenital anomalies were more common indications for testing among deletion patients, they did not reach statistical significance.

Distribution of clinical indications at the time of referral to array comparative genomic hybridisation (aCGH) testing among patients with 16p11.2 deletion and duplication. DYSMOR, dysmorphism; FTT, failure to thrive; MCA, congenital anomalies; MR/DD, mental retardation/developmental delay; SZ, seizures.
Examples of the deletions and duplications detected using a custom designed 16p-specific oligonucleotide array are shown in figure 2. We analysed 12 deletions and seven duplications out of 27 samples that comprise our cohort study. All the deletions and duplications in this region appear to be recurrent and reciprocal (minimum size 579 kb; coordinates on chr16: 29 528 190–30 107 184). After IRB approval we also analysed 30 samples (19 additional deletions and 11 additional duplications) from patients who were not recruited to our cohort as anonymised subjects, and all except for three samples showed the typical size and pattern of the 16p11.2 rearrangement presented above. One sample showed a larger size duplication (coordinate chr16: 27 283 579–31 893 687). Two of the anonymised samples showed mosaicism in the aCGH (figure 2). In one sample, a mosaic deletion was confirmed by FISH analysis which revealed that 46 of 200 (23%) interphase nuclei have the deletion. In another sample, both custom aCGH and CMA showed a log2 ratio consistent with mosaic duplication and the FISH data revealed 61% of interphase nuclei (n=200) had the duplication.

Representative Agilent 8X15K oligoarray comparative genomic hybridisation (CGH) results for heterozygous deletion, heterozygous duplication, and mosaic deletion of the proximal 16p11.2 region. Shown above is the genomic region harbouring the recurrent duplication/deletion. Red and blue bars depict low copy repeats (LCRs) flanking the recurrent rearrangement region. The representative array data below are drawn to the scale of the above region. Log2 ratio values for all oligos are plotted as a function of their chromosomal position. Probes with Log2 ratio greater than 0.25 are shown in red points, less than −0.25 are shown as green points, between −0.25 and 0.25 are shown as black points. The horizontal line for each array CGH represents an ∼10 kb moving average. The upper two arrays show a moving average of close to −1 and 0.5 in the disease locus, respectively, and therefore interpreted as heterozygous deletion and duplication. The bottom array shows a patient whose array CGH on genomic DNA from blood that reveals a moving average below, but very close to zero, therefore interpreted as mosaic deletion.
Among patients with deletion, both parents were available for FISH analysis in 10 families, and analysis of the mother alone was possible in three additional families. The deletion was confirmed to be de novo in 8/10 families, and was inherited from an affected father (08-DEL) or an asymptomatic healthy parent (mother of 09-DEL had no dysmorphic features, behavioural or cognitive problems) in one family each. Both parents were available for duplication testing in only five families, and analysis of the mother alone was possible in a sixth. Three duplications were de novo and two were inherited, one from a slightly dysmorphic and microcephalic mother (3-DUP), and the other from a cognitively impaired and microcephalic mother (6-DUP).
Deletions or duplications within this region were not observed in 194 normal parental samples analysed by aCGH, indicating that these 16p11.2 deletion and duplication CNVs are not likely to be copy number polymorphisms. These parental samples were from parents in whom a prenatal or a postnatal microarray detected CNVs in their fetus or child. Some of the parents had the same CNV detected in the proband but the parents of 16p11.2 patients were excluded.
We did not identify any known CNVs (http://genome.ucsc.edu; http://projects.tcag.ca/variation/) within the rearranged segment in our patients.
Phenotypic characterisation of patients with deletions or duplications of 16p11.2
The demographic data and the clinical characteristics of 16 patients with 16p11.2 deletions and 10 patients with the duplications are summarised in supplementary tables 1S–3S (supplementary material). One asymptomatic deletion patient (mother of 09-DEL) was not included in the statistical analysis.
The patients with deletion were younger when first diagnosed as compared to patients with duplication, but the difference was not statistically significant (two tailed p<0.08). The mean age of diagnosis was 5.9±9.1 years (range 2 months–38 years; median 2.9 years) for the deletion cases and 12.6±9.3 years (range 4–31 years; median 10 years) for duplication cases. The male to female ratio was 3.2:1 among deletion patients and 1.5:1 among duplication cases. The maternal and paternal ages did not show any biased distribution. All individuals with deletion or duplication were born appropriate for gestational age (AGA), except for 09-DUP, who was large for gestational age. The vast majority of patients with either deletion or duplication were born via spontaneous vaginal delivery with an unremarkable gestational course. Three out of 16 deletion patients needed mechanical ventilation after delivery. Other postnatal morbidities included jaundice, polycythaemia, hypoglycaemia, and temperature instability. We observed overrepresentation of the Hispanic ethnicity in our cohort (6/16 in deletion cases and 8/8 duplication cases have biparental or uniparental Hispanic background).
The common clinical manifestations associated with the 16p11.2 duplications and deletions are shown in figure 3. Speech/language delay and cognitive impairment were characteristic findings of patients with 16p11.2 rearrangements; 14/14 of the 16p11.2 deletion patients had speech/language delay with or without varying degrees of cognitive impairment. The two youngest patients in this group (9-DEL and 15-DEL) were too young to be tested for these skills. All 10 patients with the 16p11.2 duplications had speech/language delay and a more severe degree of MR when compared to the deletion cases. The one exception was the mother of patient 2-DUP who has normal speech and cognition. In addition, half of the deletion patients and 6/10 of individuals with duplication had motor delay, with the majority being in the mild range in the deletion cases. Furthermore, half of the deletion cases had mild feeding difficulties (including gastro-oesophageal reflux) during the first few weeks to months of life.

Clinical phenotypes in 16 patients with the 16p11.2 deletions and 10 patients with the 16p11.2 duplication. (see figure 2 legend for abbreviations).
Although the subjects with 16p11.2 deletion and duplication do not clearly constitute clinically recognisable syndromes, we found that certain facial features are shared among them (figures and 5). Subjects with the deletions shared the following features: broad forehead, micrognathia, hypertelorism, and a flat midface. The broad forehead, macrocephaly (see below) and flat midface give these patients a distinct facial gestalt. We found that the duplication cases were more grossly dysmorphic as compared to the deletion patients, but there was no recognisable pattern for these features.

Facial features of individuals with 16p11.2 deletion. a: case 1; b: case 2; c: case 3; d: case 4; e: case 5; f: cases 6; g: case 7; h: case 8; i: case 9; j: case 10; k: case 11; l: case 12; m: case 13; n: case 14; o: case 16. All patients with deletion showed a broad forehead and flat midface. Hypertelorism and micrognathia appear to be common findings in the 16p11.2 deletions.

Facial features of individuals with 16p11.2 duplications. a: case 1; b: case 2; c: case 3; d: case 4; e: case 5; f: cases 6; g: case 7; h: case 8; i: case 9.
Although the 16p11.2 rearrangements were initially associated with autism spectrum disorders (ASD),5 24 25 we found that only 3/16 patients with the 16p11.2 deletion meet the criteria for autism and only two patients with duplications had autistic features. On the other hand, we observed an increased incidence of other behavioural problems; six out of 10 patients with 16p11.2 duplication and six out of 16 with deletion have other behavioural abnormalities, most commonly ADHD.
Five of 16 and 3/10 patients with the deletion and duplication, respectively, had seizures, and one additional patient with a deletion had non-convulsive epilepsy. The seizures typically start during the first year of life, are easily controlled with antiepileptic medications, and tend to resolve or decrease in severity during childhood. Magnetic resonance imaging (MRI) or computed tomography (CT) studies of the brain were abnormal in seven out of 10 deletion patients who underwent this imaging because of absolute or relative macrocephaly and developmental delay. We observed an increased incidence of congenital anomalies in individuals with the 16p11.2 deletion (5/16) or duplication (5/10) with no predilection for a specific organ or system (Supplementary material). The congenital anomalies among subjects with deletion included congenital diaphragmatic hernia, chordae, cleft palate, polydactyly, congenital heart defect, multicystic dysplastic kidney, fusion of lower ribs, and pyloric stenosis. Among patients with duplication, we observed hypospadias, cleft palate, pectus excavatum, pectus carinatum, torticollis, cleft lip and palate, phimosis, pes planus, mild scoliosis, and tethered cord. Three of 16 deletion and 2/10 duplication patients had myopia and other accommodation problems.
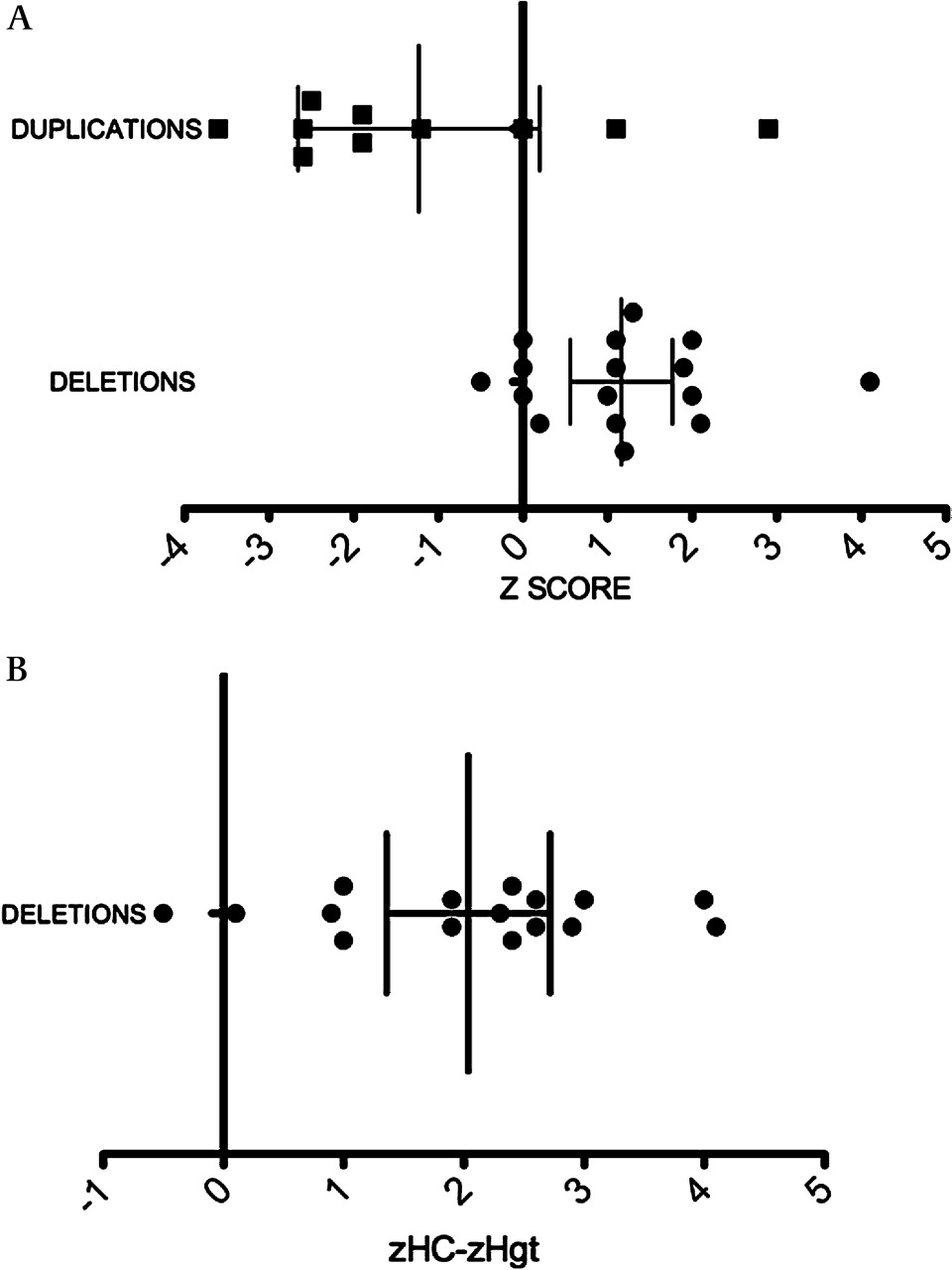
Patients with the 16p11.2 deletion or duplication had a statistically significant difference in their mean head sizes (unpaired Student t test, p<0.0017). The mean Z score for the deletion cases was +1.16 (95% CI +0.61 to +1.71), significantly different from the population mean (zero) (one sample t test, p<0.0009). Because we noted that many deletion patients had small body size, we adjusted the head circumference to the height. The mean (zHC-zHgt) score for the deletion cases was +2.025 (95% CI +1.4 to +2.65), which was also significantly different from the population mean (one sample t test, p<0.0001) (figure 6). Five deletion patients had absolute macrocephaly and six patients had relative macrocephaly. Among patients with the 16p11.2 duplication, there was a greater variation in the head size with a mean zHC of −1.23 (95% CI −2.65 to +0.19), but this did not reach statistical significance when compared to the population mean (one sample t test, p=0.08). Nevertheless, 6/10 duplication cases were microcephalic.

Deletions and duplications of 16p11.2 are associated with an abnormal head size. (A) The Z score of the head circumference among deletion (circles) and duplication (squares) patients. (B) The head size (HC) adjusted to height (Hgt). The mean of zHC-zHgt is depicted among deletion cases. All measurements are plotted as age and sex matched Z scores. Bars indicate mean and 95% CIs. Age and sex matched HC and Hgt centiles were obtained using Abase, a PalmOS-based calculator, and converted into Z scores.
Structural characterisation of LCRs and CNVs in the proximal 16p11.2 region
Detailed analyses of the rearranged 16p11.2 chromosomal region revealed that two major LCR families contribute to the complexity in this region. Two ∼147 kb segments (147A and 147B) flanking the disease locus are directly repeated and share 99.6% identity (figure 7). Immediately adjacent and distal to the 147B repeat, there is a ∼72 kb region (72C) sharing 98.6% similarity with two counterparts (72A and 72B) within the 147A and 147B LCRs and in direct orientation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the proximal 16p11.2 region based on the March 2006 freeze of the reference human genome sequence (NCBI build 36.1) and summary of genomic structure analyses. (a) Genes within this region are represented by grey bars, drawn to scale. (b) There are two major low copy repeat (LCR) families in this region. The blue arrows, termed 147A and 147B, represent the two ∼147 kb LCRs. They are in direct orientation and share 99.6% identity. The red arrows, termed 72A, 72B, and 72C, represent the three ∼72 kb LCRs. They are all in direct orientation and share ∼98.6% identity. (c) Structural variation in this region based on fosmid sequencing data from Kidd et al25 Fos1, Fos2, and Fos3 represent three groups of discordant fosmids, whose mapping is too large relative to the reference genome. The black bars at the ends indicate mapping positions of sequenced fosmid ends in the reference genome. The exact sites of structural variations are unknown because most of these fosmids are only sequenced at the ends. ABC12-J13 represents the deletion structural variation found in the fosmid ABC12-46789100J13. The green bar indicates the region deleted from this fosmid relative to the reference genome.
To investigate potential structural variation polymorphism of this region in the population, we analysed the fosmid sequencing data from Kidd et al.32 Interestingly, we found many discordant clone groups around the LCR region that, when placed on the human reference haploid genome, spanned too large a distance relative to the reference genome. This suggests potential sites of deletion polymorphism, or sites where the reference genome sequence does not represent the majority of the population. Eight/four out of nine individuals analysed by Kidd et al showed discordant fosmids similar to Fos1/Fos2 (figure 7). This suggests that an additional copy of the 147 kb LCR may exist in the population. Four out of nine individuals showed discordant fosmids similar to Fos3. One of these fosmids from the group Fos3: ABC12-46789100J13, is completely sequenced. Analysis of the sequence of this fosmid clearly shows that this individual may carry a haplotype with a deletion between 74B and 74C (figure 7). Individuals with this haplotype have: (1) shorter homologous direct LCRs flanking the disease locus; or (2) less complexity (only part of 147 kb repeat remains) compared to the reference genome (147 kb repeat plus 74 kb repeat). We hypothesise that this potential deletion polymorphism may affect an individual's susceptibility to rearrangements at this disease locus.
Discussion
We describe herein the phenotypic and molecular findings in 27 subjects with either deletion or duplication of the apparently identical genomic region within chromosome 16p11.2. Submicroscopic 16p11.2 chromosomal rearrangements are increasingly recognised as one of the most common genomic disorders, and were found in 0.6% of samples that were submitted to the diagnostic laboratory at BCM for various indications. Although initial reports described an association of 16p11.2 chromosomal rearrangements with autism, we observed a broad spectrum of clinical manifestations, and the autism phenotype was observed in only one fifth of the deletion cases. The most common clinical manifestations in our cohort were language delay and MR. The role of this genomic interval in speech/language development was also suggested in a study of Icelandic samples in which the deletion frequency was ∼0.1% among patients with a psychiatric or language disorder, as compared to 0.01% in the general population.25 Other less common phenotypes in our cohort included motor delay, seizures, behavioural problems (especially ADHD), and congenital anomalies.
The increased incidence of congenital anomalies in individuals with the 16p11.2 deletion or duplication is intriguing. Of special interest is the apparently high incidence of pyloric stenosis among patients with the 16p11.2 deletion. In addition to patient 2-DEL in our cohort, Bijlsma et al reported two patients with the same finding.26 In our cohort, we identified a high incidence of structural brain abnormalities among patients with deletions, a fact that has implications for establishing the diagnostic and surveillance guidelines for patients with the 16p11.2 rearrangements. The dysmorphic features were not emphasised in previous reports but careful physical examination revealed a high prevalence in our cohort of these abnormal findings that were more severe in individuals with the duplication. The broad forehead, macrocephaly, and flat midface render a distinct facial gestalt to the deletion patients (figure 4).
We found differences in the rate of the clinical manifestations between the deletion and duplication cases (figure 3, table 1). However, because of the small sample size of the duplication none of these differences reach statistical significance. In addition to the variable expressivity in the 16p11.2 rearrangements, we observed incomplete penetrance for the entire phenotype (mother of 9-DEL) or for the cognitive phenotype (3-DUP). The deletion cases were referred for testing 1.5-fold more frequently than the duplication cases (27 vs 18), suggesting a potential lower penetrance and/or milder expressivity of the duplications. However, this over representation of the deletions can be biologically based on the 2:1 deletion to duplication ratio underlying many known genomic disorders associated with NAHR events.33 Unexpectedly, the male to female ratio in our cohort was 3.2:1 and 1.5:1 among patients with deletion or duplication, respectively. This higher preponderance of males was also observed in all previous 16p11.2 studies (reviewed in reference 26), and is a recognised bias in classic autism.
Clinical and demographic findings in patients with 16p11.2 deletions and duplications
The microcephaly in duplication patients and macrocephaly in deletion patients, as well as the higher incidence of ADHD in the duplication patients and the presence of autism in the deletion patients, support the model of behavioural phenotypes in genomic sister disorders suggested by Crespi et al.34 35 According to this model, diametric copy number alterations can generate contrasting phenotypes associated with autistic spectrum and psychotic spectrum conditions that may represent evolution of the social brain.35 36 We observed opposing phenotypes for head circumference—that is, macrocephaly for 16p11.2 deletion and microcephaly for 16p11.2 duplication that are reciprocal to those observed for 1q21.1 rearrangements. Interestingly, at the 1q21.1 deletion/duplication locus the deletion is associated with microcephaly whereas the duplication is associated with macrocephaly.23 Deletion of 1q21.1 is also associated with schizophrenia.18 19 However, we do not observe other opposing phenotypes for duplication versus deletion patients (table 1).
The differences in the mean head sizes in patients with the 16p11.2 deletion and duplication as compared to controls were striking. The mean head sizes of deletion patients were above normal, and 2/3 of them had absolute or relative macrocephaly (figure 6). The duplication patients had small mean head sizes with 6/10 having microcephaly (figure 6). Interestingly, macrocephaly is found in a high frequency in patients with autism spectrum disorders, and small head sizes are overrepresented in psychotic spectrum conditions.35 These observations raise intriguing questions regarding the evolutionary aspects of the human cognitive architecture and the development of the social brain in humans.
The analysis of the genomic structure of the rearranged 16p11.2 chromosomal region revealed two major LCR families (147 kb and 72 kb repeats) that contribute to the complexity in this region (figure 7). These repeats are most likely responsible for generating the observed recurrent deletions/duplications by NAHR in our cohort. However, further studies to refine the recombination hotspots are required to determine precisely which LCRs are involved in these chromosomal rearrangements. We also performed in silico analyses to investigate potential structural variation polymorphisms in this region and identified a deletion polymorphism that might affect an individual's susceptibility to rearrangements in the disease locus (figure 7).
In the three previous 16p11.2 studies the great majority of deletions were de novo.5 23 24 A more recent study reported six familial cases among 14 index patients.26 In this study, the transmitting parent had developmental problems in half of the familial case.26 In our cohort, the chromosomal rearrangements were de novo in most of the deletion cases in which parental studies were available, but inherited and de novo patterns were observed in the subjects with duplications. The reduced penetrance and variable expressivity in these studies on the 16p11.2 rearrangements are important findings for genetic counselling of newly diagnosed patients and for antenatal diagnoses. Variability in the phenotype has also been observed in other genomic disorders such as the Smith Magenis microdeletion syndrome (MIM 182290) despite a common sized genomic imbalance.37 Some variability may relate to SNPs or CNVs on the remaining hemizygous allele.38 Perhaps mosaicism for this frequent rearrangement might potentially contribute to both penetrance and variable expressivity, but additional patients need to be evaluated and further molecular studies are required to clarify this point. No imprinted genes have been identified so far within the rearranged region (according to the http://geneimprint.com/website, October 2009), suggesting that imprinting is unlikely to modify the phenotype or contribute to the phenotypic variability of these individuals.
Our study clearly demonstrates a dosage effect of 16p11.2 copy number on the various clinical findings, and suggests the presence of dosage sensitive genes within the rearranged interval. It is important to note that, in addition to causing autosomal dominant phenotypes, the deletion of genes can occasionally unmask a mutation in the second allele, resulting in an autosomal recessive phenotype, or could cause an imprinting disorder due to deletion of imprinted genes.39 This clinical scenario has been recently described in a patient with severe combined immunodeficiency due to Coronin-1A (MIM 605000) mutation and a 16p11.2 deletion.40 The rearranged 16p11.2 interval contains 27 annotated genes (figure 7), and some are promising candidates for the different phenotypes in patients with these syndromes. MAPK3 (MIM 601795) is a synaptic signalling component necessary for several forms of learning. Mapk3−/− null mice showed a dramatic enhancement of striatum dependent long term memory.41 The TBX6 (MIM 602427) gene encodes a transcriptional regulator involved in developmental processes and can play a role in the congenital anomalies that were observed in our patients. SEZ6L2 is an apparent seizure related gene with high central nervous system (CNS) specific levels of expression. Recently, a sequence variation was identified in SEZ6L2 that may represent a novel genetic risk factor for autism.42 The QPRT (MIM 606248) encodes quinolinate phosphoribosyltransferase, a key enzyme in catabolism of a potent endogenous exitotoxin to neurons called quinolinate. Elevation of quinolinate levels in the brain due to decreased activity of this enzyme has been linked to the pathogenesis of epilepsy in humans.43 The DOC2A (MIM 604567) encodes a protein predominantly expressed in brain and is possibly involved in calcium dependent neurotransmitter release and in dynein dependent intracellular vesicle transport.44 The protein serine/threonine phosphatase 4 encoded by PPP4C (602035) interacts with the survival of motor neurons complex45 and is regulated by histone deacetylase 3.46 The MAZ gene (MIM 600999), expressed in human embryonic brain and in mouse brain, encodes the transcription factor MYC associated zinc finger protein which enhances the NMDA receptor subunit type 1 (MIM 138249) activity during neuronal differentiation.47 In addition, overexpression of MAZ can inhibit cell cycle proliferation in rabbit synoviocyte cells.48
In conclusion, our results expand the spectrum of phenotypic abnormalities observed in patients with the 16p11.2 deletion or duplication and reveal specific objective clinical features (eg, microcephaly or macrocephaly) associated with copy number imbalances at this locus. These deletion/duplication genomic disorders are highly associated with speech/language delay, cognitive impairment, and abnormal head size. Motor delay, neuropsychiatric abnormalities, congenital anomalies, and seizures are also frequent phenotypic findings. The incomplete penetrance and variable expressivity of clinical findings in patients with these rearrangements complicates both the clinical interpretation of the molecular data and the genetic counselling. The phenotypic variability may be related to other genetic or genomic variants, but further molecular analysis is essential to enable robust genomotype/phenotype correlations.
Acknowledgments
The authors thank the patients and parents for their willingness to participate in our research study. We thank Dr Feng Zhang for assistance in the design of the high-resolution 16p11.2 specific array. P.S. was supported in part by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education. This work was supported in part by National Institute of Neurological Disorders and Stroke, N. I. H. grant RO1 NS058529 to J.R.L.
Appendix
While this manuscript was in review a paper appeared that associated duplication of 16p11.2 with schizophrenia in adult patients: McCarthy, et al (2009) Microduplications of 16p11.2 are associated with schizophrenia. Nature Genetics doi: 10.1038/ng.474.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Competing interests Declaration: Some authors are based in the Department of Molecular and Human Genetics at Baylor College of Medicine (BCM), which offers extensive genetic laboratory testing, including use of arrays for genomic copy number analysis, and derives revenue from this activity. JRL is a consultant for Athena Diagnostics, 23 and Me, and Ion Torrent Systems.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the IRB-Baylor College of Medicine, Houston, Texas, USA.
Provenance and peer review Not commissioned; externally peer reviewed.