Article Text

Abstract
Background Williams–Beuren syndrome (WBS) is a developmental disorder with multisystemic manifestations mainly characterised by vascular stenoses, distinctive craniofacial features, mental retardation with a characteristic neurocognitive profile, and some endocrine and connective tissue abnormalities, caused by a recurrent deletion of 1.55 Mb including 26–28 genes at chromosomal region 7q11.23. The analysis of clinical–molecular correlations in a few reported atypical patients has been useful to propose several deleted genes as main contributors to specific aspects of the WBS phenotype.
Patients and methods Two additional families with partial phenotypes and atypical 7q11.23 deletions were studied. Deletions were precisely defined at the nucleotide level, and the expression levels of some affected and flanking genes were assessed in lymphoblastoid cell lines.
Results Affected individuals presented variable cardiovascular and connective tissue manifestations, subtle craniofacial features, normal visuospatial construction abilities with low average IQ and no endocrine abnormalities. The deletion in family NW1 encompassed 817 kb with 11 genes (CLDN3–GTF2IRD1), and 610 kb with 14 genes (VPS37D–RFC2) in family NW2. All deleted genes in typical and atypical deletions revealed low expression levels in lymphoblastoid cell lines, except for GTF2IRD1. CLIP2 was also underexpressed in all patients despite being outside the deletion in NW2, while no other flanking non-deleted gene showed significantly different expression compared to controls.
Conclusions Along with previously reported cases, clinical–molecular correlations in these two families further confirm that the functional hemizygosity for the GTF2I and GTF2IRD1 genes is the main cause of the neurocognitive profile and some aspects of the gestalt phenotype of WBS.
- Williams–Beuren syndrome (WBS)
- partial deletion
- visuospatial construction
- mental retardation
- genetics
- clinical genetics
- cytogenetics
- molecular genetics
- visual development
Statistics from Altmetric.com
- Williams–Beuren syndrome (WBS)
- partial deletion
- visuospatial construction
- mental retardation
- genetics
- clinical genetics
- cytogenetics
- molecular genetics
- visual development
Williams–Beuren syndrome (WBS, Mendelian Inheritance in Man 194050) is a rare neurodevelopmental disorder with a prevalence of 1/7500 to 1/15 000 live births. WBS individuals present with typical dysmorphic features, mild to moderate mental retardation and a characteristic cognitive profile that includes relatively preserved verbal skills but very deficient visuospatial abilities. They also have characteristic personality traits, such as preferring the company of adults to their peers and lacking shyness or fear with strangers. Most of them have short stature, cardiovascular abnormalities (typically supravalvular aortic stenosis (SVAS) and peripheral pulmonary stenosis (PPS)), hyperacusis, transient infantile hypercalcaemia and glucose intolerance as adults.1–4 WBS is caused in most cases by a 1.55- to 1.83-Mb deletion at chromosomal region 7q11.23, including 26–28 annotated genes depending on the deletion breakpoint.5–13 Similar-sized deletions occur in a recurrent manner and with a relatively high mutation rate due to the regional genomic architecture with flanking segmental duplications that predispose to non-allelic meiotic recombination.14 However, atypical rearrangements are non-recurrent in general and only account for 2% to 3% of cases.
The first gene mapped to the WBS deleted interval that was linked to a specific phenotype was the gene coding for elastin (ELN). Deletion, disruption or mutation of ELN causes the cardiovascular and connective tissue phenotype of WBS, including SVAS.15–18 A few atypical patients with deletions that do not span the entire WBS region have been reported so far. Clinical–molecular correlations in these patients provide a unique opportunity to investigate the individual contribution of genes within the hemizygous interval to the WBS phenotype.15 19–30 Based on two families presenting with cardiovascular manifestations and partial features of the WBS cognitive profile associated with small deletions involving only ELN and the LIM kinase-1 coding gene (LIMK1), hemizygosity of LIMK1 was proposed as a contributing factor to impaired visuospatial constructive cognition in WBS.19 However, this claim is not consistent with the finding on different patients with similar or larger heterozygous deletions, including LIMK1, who also showed SVAS but no facial gestalt and no cognitive deficits, lacking the typical spatial and constructive impairment on repeated testing.20 21 In addition, several patients with deletions spanning from around the centromeric common breakpoint of the WBS typical deletion to near the CLIP2 gene have been described in individuals presenting with mild or no mental retardation, preserved visuospatial cognition and mild facial anomalies.22–25 30 This would imply that genes affected by deletions other than ELN (from NSUN5 to EIFH4) appear to contribute modestly to the WBS phenotype and little to neurocognition. A patient with typical facial features without the typical neurocognitive profile, only mild visuospatial construction deficit, was found to have a similar deletion but with the telomeric breakpoint within GTF2IRD1.27 Moreover, five families with deletions spanning almost the entire interval (from FZD9 to GTF2IRD1) had visuospatial constructive cognition impairment but no global mental retardation.26 The description of two patients with the full WBS phenotype who have deletions from ELN to GTF2I supports the hypothesis that the genes mainly responsible for abnormal cognition map to the telomeric interval of the deletion (CLIP2, GTF2IRD1 and GTF2I).28 29 Atypical deletions that span part of the WBS critical region extending towards the telomere or centromere have also been reported in patients with more complex phenotypes.31–33
Despite these relatively consistent clinical–molecular correlations, the number of patients with atypical deletions is rather small, and detailed deletion mapping at the sequence level and expression data of deleted and flanking genes are missing in most reported cases. Other features of the WBS phenotype, such as the metabolic abnormalities, have not been associated with specific genes. Here, we report on two new families with fully characterised atypical 7q11.23 deletions and partial phenotypes. The combination of this information with the already published atypical deletion cases allows us to further refine the genotype–phenotype correlations in WBS patients.
Materials and methods
Subjects/medical evaluation
Patients were initially ascertained in the Centro de Salud Azahara, Córdoba, Spain (family NW1), and in the Medizinische Universität Innsbruck, Austria (family NW2). In addition to a complete medical check-up following the standard protocol for WBS patients, intellectual performance of the adults in both families was evaluated using the WAIS III or WIE (Spanish and German versions of the Wechsler Adult Intelligence Scale),34 and visuospatial skills were tested using the block design subtest of WAIS III or WIE, developmental test of visual–motor integration and Rey visual design learning test.21 Other abilities were also evaluated, such as attention, memory, executive functions, fine-motor coordination and calculation.35 36 Infants in the NW1 family were evaluated using the Denver developmental scale.
Sample preparation
Blood samples were obtained from patients and available first-degree relatives under informed consent approved by the institutional review board. Genomic DNA was extracted directly from peripheral blood using the PureGene kit (Gentra Systems, Minneapolis, Minnesota, USA) according to the manufacturer's instructions. Epstein–Barr virus-transformed lymphoblastoid cell lines (LCLs) were established from an affected individual from each family (individual NW1 III.4 and individual NW2 II.1). We also used LCLs from four individuals with WBS and typical deletions, as well as several Spanish unrelated individuals as controls. LCLs were grown in RPMI 1640 medium (GIBCO, Invitrogen, Carlsbad, California, USA) with 10% foetal calf serum and antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin) at 37°C with 5% CO2.
Genomic DNA and total RNA were extracted from each cell line with the PureGene kit (Gentra Systems) and with TRIZOL reagent (Invitrogen), respectively, according to the manufacturer's instructions. The concentration and quality of all DNA and RNA samples was checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California, USA). Total RNA was converted to cDNA using SuperScript II (Invitrogen) reverse transcription primed with random hexamers.
Multiple-ligation probe amplification and microsatellite analyses
A custom-made multiple-ligation probe amplification (MLPA) panel, with specific oligonucleotide probes for genes located in the commonly deleted interval and flanking regions, was designed (Supplementary table 1). All gene probes (n=16) were tested in a single assay using 150 ng of genomic DNA. The resulting fragments were separated by capillary electrophoresis in a sequencer (ABIPrism 3100, Applied Biosystems, Foster City, California, USA ). Samples were analysed by visual examination of the peak profiles and by exporting the peak heights to an Excel sheet with data calculation as previously described.37
PCR analyses of single-copy and multiple-copy microsatellites mapping to the 7q11.23 region were also used to define the parental origin of the rearrangement. PCR conditions and oligonucleotide primers were as previously described in detail.5
Comparative genomic hybridisation in microarrays and junction fragment mapping
Microarray comparative genomic hybridisation analysis was carried out using the microarray analysis platform of NimbleGen Technologies (Roche NimbleGen, Madison, Wisconsin, USA). Patient and reference genomic DNA samples were independently labelled with fluorescent dyes, cohybridised to a NimblenGen Human Chromosome 7 Tiling Array. Analysis of microarray data was performed as described by the manufacturers.38 For each spot on the array, log 2 ratios of the Cy3-labelled test sample versus the Cy5 reference sample were calculated. Corrected ratios above 0.2 and below −0.2 were considered as deletion or gain. Window averaging was performed with 4000 base pairs (bp), 8000 bp and 20 000 bp, and the region of interest was visually screened with SignalMap v.1.9 from NimbleGen. Based on the information obtained, we designed PCR primers to amplify the deletion junction fragment (Supplementary table 2), using the Expand Long Template Taq polymerase (Roche, Mannheim, Germany) and standard conditions. Sequencing of the PCR product was performed in an ABI 7900 HT sequencer (Applied Biosystems) with Big Dye 3.1 kit.
Real-time quantitative reverse transcription-PCR and data analysis
We used UPL probes (Roche) and TaqMan Universal PCR Master Mix 2X (Applied Biosystems) or SYBR green master mix. Primers and probes were designed using the ProbeFinder software version 2.34 from Roche, with default parameters. In all cases, oligonucleotide primers were designed to span across an intron. We designed an assay for 10 genes (primers available on Supplementary table 2).
Two genes were selected as reference for normalisation on the basis of their stability with regard to expression levels in LCLs, RAD51L1 and AGPAT1. Real-time PCR reactions were set up in a 10 μl volume in a 384-well plate with three replicates per sample. All PCR products were run in an ABI 7900 Sequence Detection System (Applied Biosystems) with the following conditions: 50°C for 2 min, 95°C for 10 min and 40 cycles of 95°C 15 s/60°C for 1 min. Raw data were obtained using SDS 2.1 (Applied Biosystems). Subsequent assay efficiency calculations and measurements of relative expression were carried out in Excel (Microsoft, Redmond, Washington, USA), applying the standard curve method. Expression values obtained from individual samples were normalised against a pool of cDNA made of five control samples, and were plotted against the average values obtained from controls (n=5) and WBS individuals (n=4). A t test comparison was performed to define whether the differences found between WBS individuals and controls were statistically significant.
Results
Clinical description
Family NW1
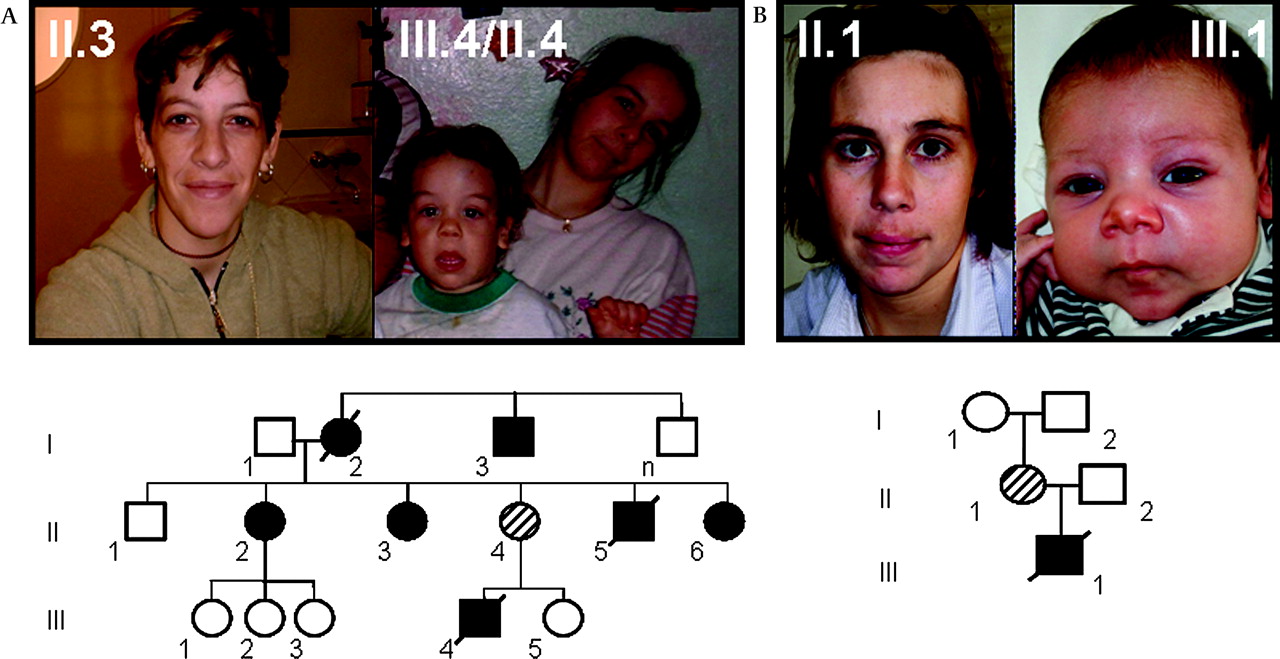
At least eight patients from a three-generation family presented with some craniofacial features reminiscent of WBS (figure 1), cardiovascular problems of variable severity, mostly SVAS, and additional connective tissue problems, as well as mild cognitive deficits.39 Anthropometric parameters and growth charts were normal in all affected individuals and no other relevant clinical manifestations related to WBS were present. Specifically, no endocrine abnormalities of calcium and glucose metabolism were detected on repeated testing, including normal glucose overload test in women during pregnancy. The grandmother (NW1 I.2) died at 35 years of age; her sudden death was of cardiovascular origin. Two other family members, child NW1 III.4 and his uncle (NW1 II.5), also died suddenly with cardiac arrest at 4 and 15 years of age, respectively. The family lived in a remarkably poor social and educational setting. Detailed clinical description of the affected individuals who were completely examined is on table 1.
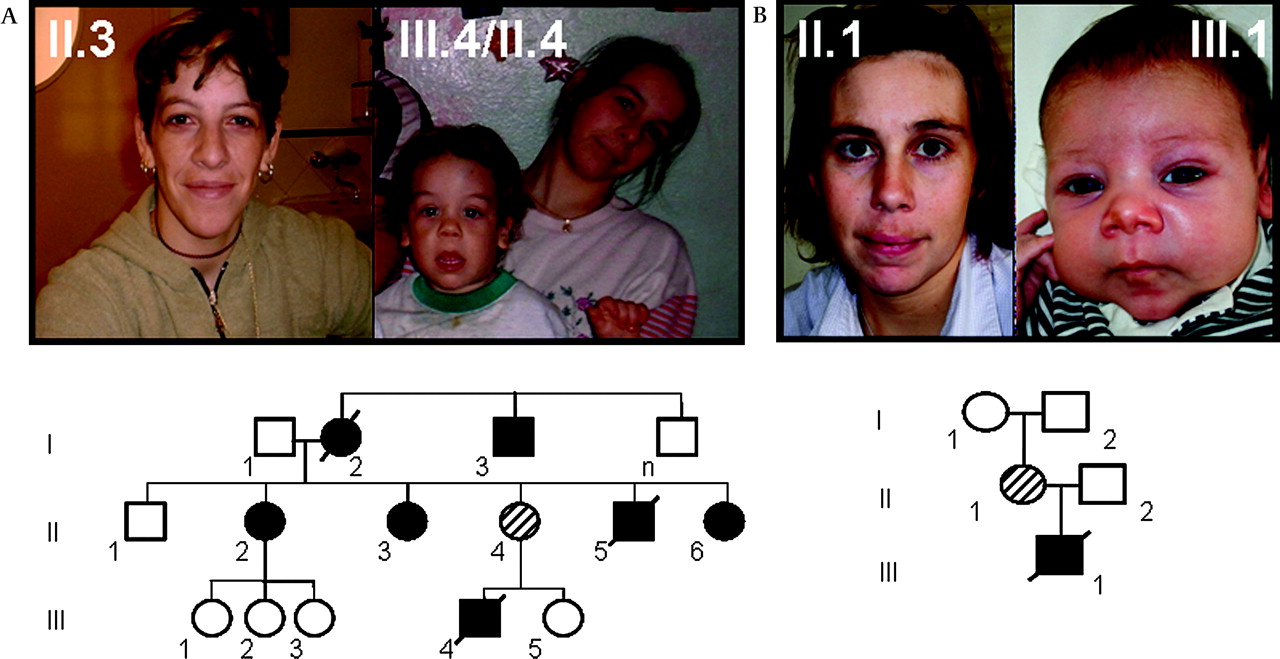
Pictures of some affected members and pedigree of the two families studied. (A) family NW1 and (B) family NW2. In family NW1, there are several affected individuals in three generations with similar phenotype except for incomplete penetrance of supravalvular aortic stenosis. In family NW2, the deletion occurred de novo in individual II.1, and the proband (III.1) died at 4 months due to cardiovascular problems. Individuals in the pictures provided consent for publication.
Clinical characteristics of atypical patients in comparison with WBS patients
Neuropsychological testing was performed on patients NW1 II.4 and II.3. They had a full-scale IQ of 70 and 80, a verbal IQ of 73 and 81, and a performance IQ of 70 and 83, respectively (figure 2). In both cases, culturally influenced items showed extremely poor results, suggesting that these results are influenced by the marginal social milieu. Moreover, fatigue and distraction due to the presence of her newborn child could have affected results in patient NW1 II.4, obtaining a lower punctuation than her real possibilities, since she is autonomous in a familial and social context. The results in all visuospatial construction tasks were strictly normal in both sisters—that is, close to average. NW1 III.4 was evaluated at 18 months of age. He showed a normal psychomotor development and was functioning at the appropriate age level in all tasks according to the Denver developmental scale.

Performance on neuropsychological background tasks on the German and Spanish versions of the Wechsler Adult Intelligence Scale on patients NW1 II.4 (light grey) and NW2 II.1 (dark grey). Please note that for the sake of simplicity, all test scores were converted into z-scores (mean 0 (SD 1)) and, consequently, average performance is reflected by z-scores between −1 and 1. Thus, a z-score below −1 reflects deficiencies (defined as 1 SD below age norm).
Family NW2
This two-generation family was ascertained after the diagnosis of a newborn baby with severe SVAS. The child died at 4 months of age due to complications following cardiac surgery for SVAS. The mother had normal cardiac evaluation by echocardiography and displayed only minor facial features of WBS at age 30 years, as well as her child at the moment of examination (figure 1). Like in family NW1, no metabolic abnormalities were detected in NW2 II.1, including normal glucose overload test during pregnancy. Her neuropsychological testing revealed a discrepancy between verbal IQ (below average range) and performance IQ (low average range), with a full-scale IQ slightly below the average range. Moreover, the index scales derived from the IQ assessment disclosed average performance in three out of four indices: working memory, perceptual organisation and language comprehension. Slightly subaverage performance was observed in the index scale measuring information-processing speed (figure 2). Importantly, performance on neuropsychological background tasks was average with respect to tasks tapping attention, learning and memory, executive functions, fine-motor coordination and visuospatial abilities (Supplementary table 3). However, consistent with the literature,21 patient NW2 II.1 experienced circumscribed difficulties on a rather easy calculation test that was designed for brain-damaged patients; specifically, she encountered severe difficulties to solve written multiplications. Detailed clinical description is on table 1.
Identification and molecular characterisation of the deletions
Family NW1
Affected family members revealed a heterozygous deletion, including the MLPA probes for the genes STX1A, LIMK1, EIF4H, RFC2, CLIP2 and GTF2IRD1, while control genes outside this region showed a normal dose. Microsatellite analysis revealed a hemizygous deletion of markers ELN I1 and GTF2IRD1-CAn, while markers D7S1870, CR16T and AFMb055×e5 showed biparental inheritance. Analysis with the chromosome 7 tiling microarray from NimbleGen allowed us to refine the deletion between genes ABHD11 and CLDN3 in the proximal breakpoint and within the GTF2IRD1 gene in the distal breakpoint. We designed primers to amplify and sequence the junction fragment in all affected individuals from whom samples were available. The deletion was 817 906 bp in size, with breakpoints located at chromosomal 7 locations 72 803 709 bp (proximal breakpoint) and 73 621 615 bp (distal breakpoint), based on human genome build 36. The proximal or centromeric breakpoint was mapped to an AluSq element, while the distal or telomeric breakpoint is found in a Mammalian-wide Interspersed Repeat (MIR) element (figure 3).
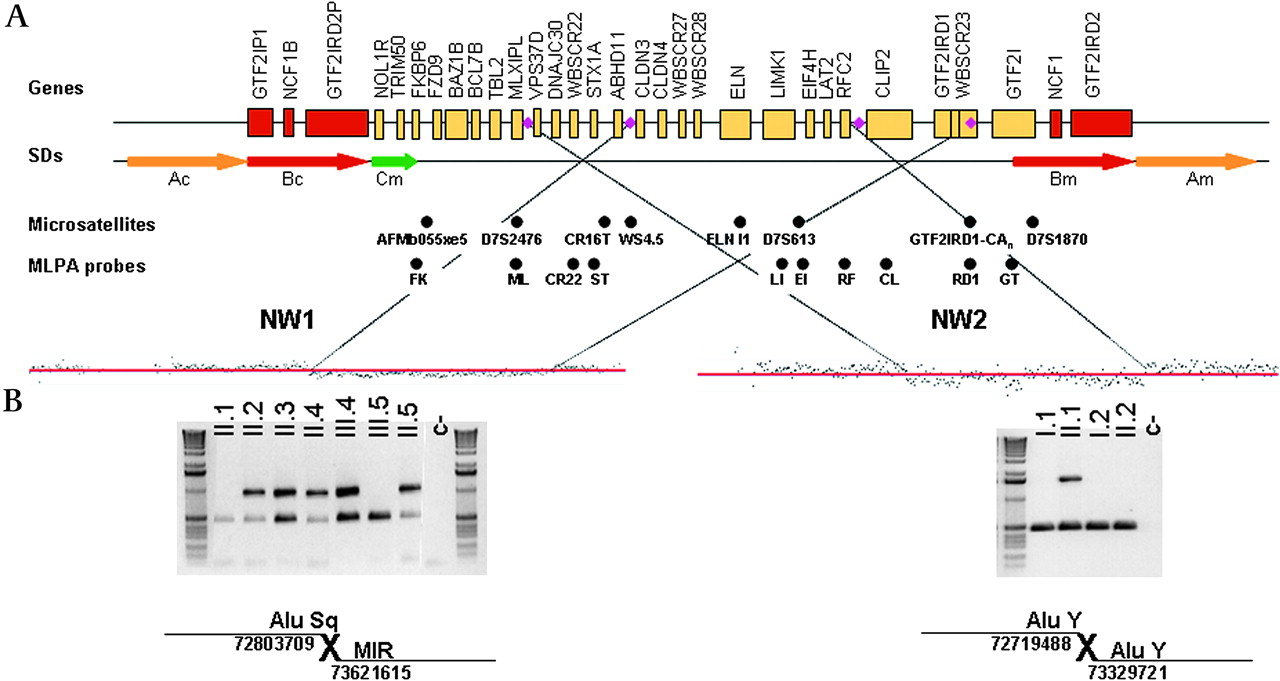
Molecular characterisation of the atypical deletions. Top, schematic representation of the 7q11.23 chromosomal region, with single copy genes (black line) and segmental duplications (SDs; arrows). Deletion breakpoints of both atypical deletions are marked with a purple rhombus. The microsatellites and MLPA (multiple-ligation amplification probe) probes analysed are shown below. (A) Mapping of deletion breakpoint by array comparative genomic hybridisation (NimbleGen Chromosome 7 Tiling Array). (B) PCR amplification of deletion junction fragments with the presence of repetitive elements.
Family NW2
The proband and his mother showed a hemizygous deletion of the MLPA probes for the genes WBSCR22, STX1A, LIMK1, EIF4H and RFC2, while gene probes outside this region showed a normal diploid dose. The microsatellites CR16T, WS4.5 and D7S613 showed a single allele and lack of maternal inheritance in the proband and his mother, while the maternal grandparents were heterozygous. Haplotype data revealed that the deletion had occurred de novo in the mother, in her maternally inherited chromosome. By high-resolution microarrays, the proximal deletion breakpoint was mapped inside the VPS37D gene and the distal breakpoint 12 kb upstream the CLIP2 gene transcription initiation site. We also amplified and sequenced the specific deletion junction fragment in both patients to precisely define the breakpoints. The deletion was 610 233 bp in size, with breakpoints at chromosomal locations 72 719 488 (proximal breakpoint) and 73 329 721 (distal breakpoint) of chromosome 7. Both breakpoints fell within the AluY sequence elements (figure 3).
Expression profile of deleted and flanking genes in the 7q11.23 region
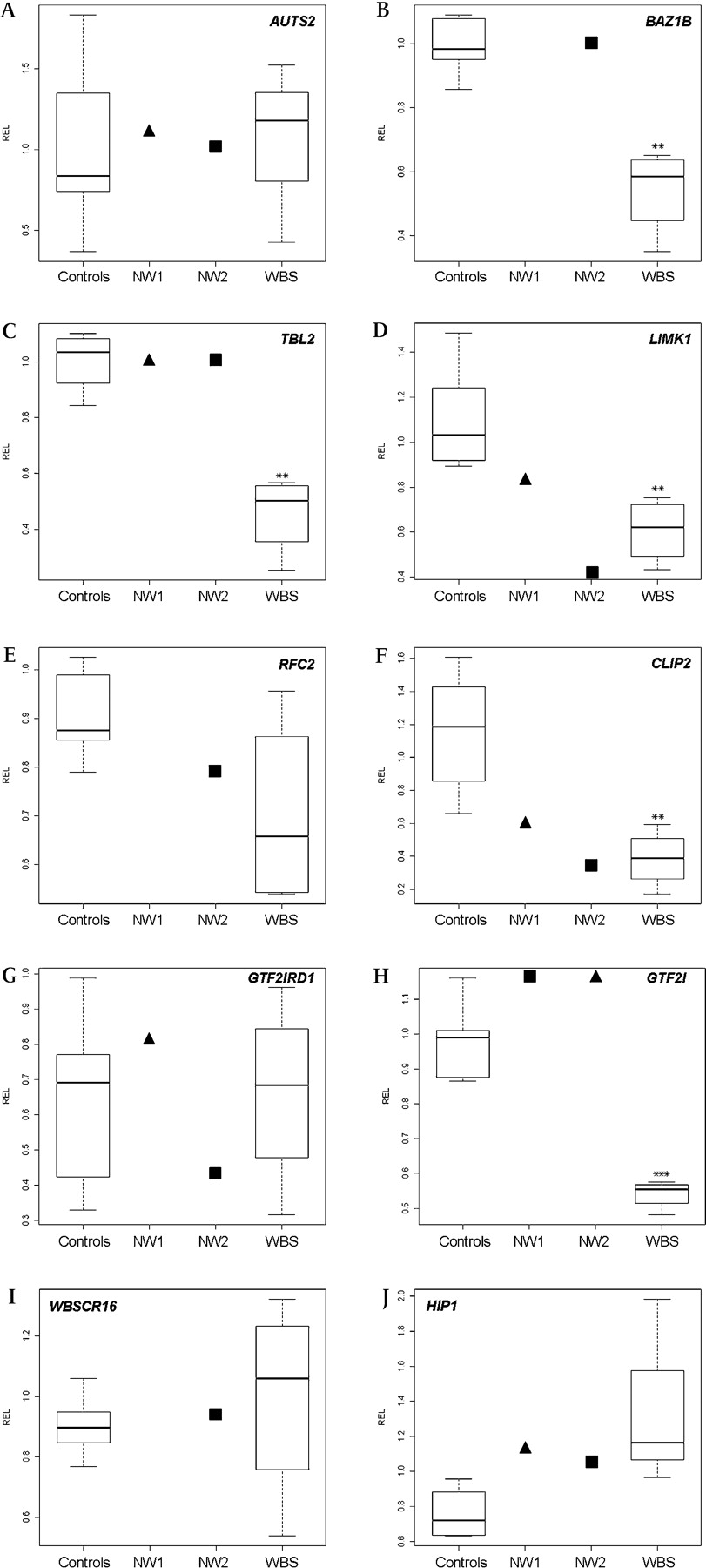
To define dosage-sensitive genes in the deleted and flanking regions, we quantified the gene expression in LCLs by real-time quantitative reverse transcription-PCR. We compared gene expression levels in our two atypical WBS patients with the common deletion (n=4) and controls (n=5). We analysed three genes located in the 1 Mb region flanking the common WBS deletion, AUTS2 (upstream) and WBSCR16 and HIP1 (downstream), outside of the deletion in all cases. None of these genes showed a significant reduction on its expression levels with respect to controls. BAZ1B, TBL2 and GTF2I (deleted in WBS patients but not in any of our two families) showed a reduction in its expression in WBS patients with respect to controls, but not in our patients, as expected. We also analysed two genes that comprised the deleted interval in both patients (LIMK1 and RFC2) and two genes deleted only in family NW1 but not in family NW2 (CLIP2 and GTF2IRD1). LIMK1 and RFC2 presented a decrease in its expression in our patients and in WBS patients. CLIP2 also showed a lower expression in all WBS patients and both our patients, despite not being deleted in family NW2. Given that the CLIP2 gene transcription initiation point is presumably located 12 kb downstream of the distal deletion breakpoint, its underexpression could be due to the deletion of a cis regulatory element or a positional effect. In contrast, we did not find a differential expression of the GTF2IRD1 gene between WBS patients and controls, as reported elsewhere,40 and its expression was also normal in our cases. See figure 4 and Supplementary table 4 for details.

Relative expression levels (RELs) of relevant genes in lymphoblastoid cell lines of four WBS patients, six controls and two individuals with atypical deletions, NW1 III.4 (black triangle) and NW2 II.1 (black square). REL boxplots are shown for 10 genes, 3 flanking the commonly deleted region (A, centromeric; I and J, telomeric) and 7 located in the commonly deleted interval (B–H). **p<0.01 and ***p<0.001, t test. No data were available for three genes in NW1 III.4 due to technical problems.
Discussion
Molecular characterisation of partial deletions: rearrangement mechanism
We have studied two new families with atypical deletions of the WBS region in 7q11.23 that are associated with partial WBS phenotypes. The deletion in family NW1 encompasses 817 kb including 11 genes (CLDN3-GTF2IRD1), while the deletion of family NW2 is 610 kb in length including 14 genes (VPS37D-RFC2) but functionally also affecting the next distal gene, CLIP2. It is worth noting that the GTF2IRD1 gene, one of the few genes differentially deleted in the two atypical deletions, does not show any differential expression when comparing LCLs from patients and controls, as previously reported.40 However, its expression could still be different in other tissues or in the developmental stages between WBS patients and controls, as already reported in skin fibroblasts.40
At the sequence level, we have precisely defined the deletion junction fragments, and we have found that the regions implicated in the rearrangement in both families contained repetitive elements. We observed in family NW1 an AluSq and a MIR element, whereas in family NW2 both breakpoints were within AluY elements. Alu sequences are thought to be frequently involved in germline and somatic genomic rearrangements.41 However, given the low level of sequence similarity between the deletion breakpoints, the most likely mechanism leading to the rearrangements is non-homologous end joining or fork stalling and template switching.42 While WBS deletions occur in large regions of high homology with specific mechanisms of abnormal chromosomal pairing and exchange, these non-recurrent deletions are likely random events that could be facilitated by other repetitive elements of the human genome.
Clinical–molecular correlations
Clinically, family NW1 presented with some craniofacial features of WBS, a cardiovascular phenotype with incomplete penetrance, outgoing personality, learning problems and borderline IQ, but no visuospatial constructive impairment. Similarly, family NW2 presented severe SVAS in only one of the two affected members (incomplete penetrance) and normal visuospatial constructive cognition along with slightly subaverage general intellectual abilities. Therefore, clinical–molecular comparisons of our patients with respect to the WBS individuals further reinforce that LIMK1 deletion alone would not be sufficient to cause the impairment of visuospatial and construction abilities as once proposed,20 and that the deletion of GTF2I and GTF2IRD1 may contribute to some of the craniofacial features, the global intellectual deficit and some aspects of the cognitive profile, such as visuospatial constructive cognition. It is worth noting that patients of the NW1 family present a deletion of GTF2IRD1 but not of GTF2I, allowing the dissection of their individual contribution to visuospatial construction deficit, personality and facial features. Our data confirm that hemizigosity for GTF2IRD1 alone is not sufficient to cause the visuospatial construction deficit but may contribute to the hypersociable personality; the contrary was observed and hypothesised in a patient with a similar deletion breakpoint on the telomeric end.27 (figure 5) In addition, some other features absent in our patients and highly prevalent in WBS, such as impaired glucose tolerance and silent diabetes that is present in 75% of adult WBS individuals,4 might be caused by differentially deleted genes, including GTF2I and GTF2IRD1, and all the genes centromeric to VPS37D. An excellent candidate for this metabolic phenotype is the MLXIPL gene, coding for the carbohydrate response element-binding protein.43 Recently, mouse models that replicate the human molecular deletion and mice with half of the region deleted have been reported.44 These partial-deletion mice replicate crucial aspects of the human disorder and serve to identify genes and gene networks contributing to the multisystemic and neurodevelopmental abnormalities of WBS.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
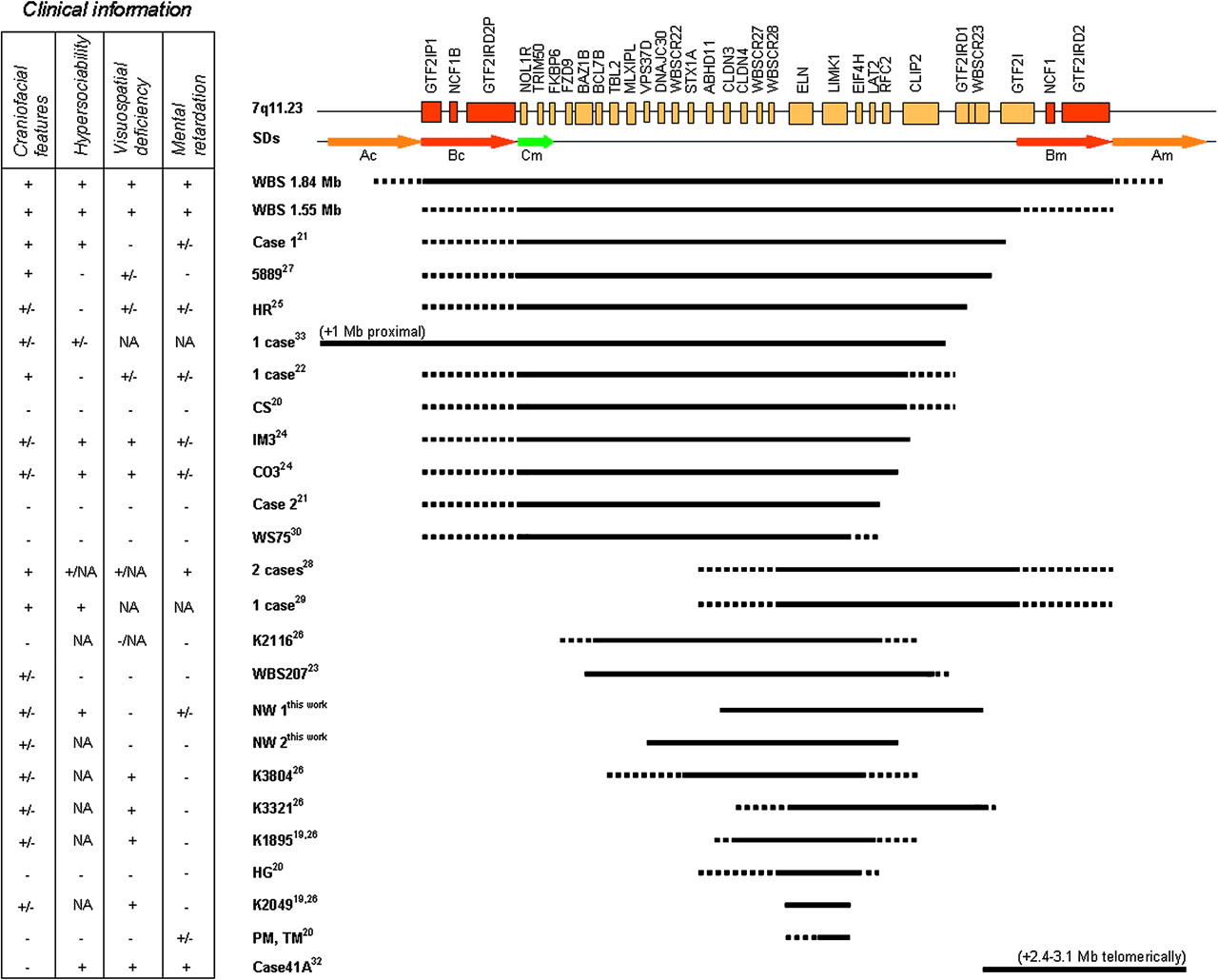
Typical and atypical deletions of the 7q11.23 region reported to date with respect to the transcript map of the Williams–Beuren syndrome (WBS) critical region. Genes are represented by yellow boxes, and arrows indicate segmental duplications flanking the deleted region (block A in yellow and block B in red). The extent of the deletion observed in classic WBS patients (1.55 and 1.84 deletions), as well as for the atypical deletions, is denoted by a dark line below the transcript map. Unclear deletions statuses are represented by dot lines. Patients' symbols and references are indicated on the right of the dark line. Clinical information in the box. +, present; +/−, ambiguous/borderline; −, not present; NA, not available.
Contribution of the TFII-I family of transcription factors to the WBS cognitive profile
Our patients add relevant information to the previously reported cases with partial deletions of the WBS locus, allowing the definition of a more precise phenotypic map of the region based on clinical–molecular correlations (figure 5). The existence of a critical region responsible for the cognitive profile, which includes the GTF2I and GTF2IRD1 genes, is becoming more evident. These genes are always deleted in WBS patients. It is worth noting that depending on the deletion breakpoint in classic WBS patients, two genes are not always deleted and could contribute to the phenotypic variability—GTF2IRD2 and NCF1. NCF1, coding for the p47phox subunit of the NADPH oxidase complex, has already been defined as a modifier for the risk of hypertension in WBS.6 GTF2I, GTF2IRD1 and GTF2IRD2 are members of the TFII-I family of proteins, characterised by the presence of multiple helix–loop–helix-like novel domains known as I-repeats.45 GTF2I and GTF2IRD1 both contain multiple I-repeats. GTF2I contains both DNA and protein binding sites and is a multifunctional transcription factor that can bind enhancer (E-box) and core promoter (Inr) elements.45 Similarly, GTF2IRD1 is also thought to have a gene-regulatory function through direct DNA interactions.46 The GTF2IRD2 gene function is still unknown, although the presence of several features characteristic of regulatory factors, including two I-repeats, two leucine zippers and a single Cys-2/His-2 zinc finger, suggests that GTF2IRD2 has DNA-binding and protein-binding properties.12 47 Due to its location, variably included in the WBS deletions, GTF2IRD2 is also a candidate to modulate the effects of the other TFII-I-related genes on the WBS phenotype.
To strongly define the genes responsible for each aspect of the WBS phenotype and their interacting effects, a detailed molecular characterisation of all patients with partial deletions should be accomplished, including expression data of regional genes along with a comprehensive and homogeneous clinical phenotyping. Moreover, mice models for specific genes of the region or deletion of half of the critical region deleted in patients are powerful tools for establishing genotype–phenotype correlations. The analysis of these two novel families along with all previously reported data strongly reinforce the knowledge that WBS is a real contiguous gene syndrome caused by the additive effects of haploinsufficiency for several genes in the 7q11.23 critical region. While ELN deletion is responsible for the cardiovascular phenotype, with NCF1 as a modifier for blood pressure, the TFII-I-related genes appear to be the major players for the neurocognitive profile, especially mental retardation and visuospatial impairment, as well as contributors to some craniofacial features of WBS.
Acknowledgments
We thank the patients and their families for their support. We also thank Ivon Cuscó for critical reading of the manuscript and Benjamí Rodríguez-Santiago for help in the elaboration of a figure. This work was supported by grants from the Spanish Ministries of Science and Education (SAF2004-6382) and Health (FISPI042016 and FISPI076832), and the VI Framework Programme of the European Commission (LSHG-CT-2006-037627).
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Clinical Research Ethics Committee from IMIM/UPF/PRBB (http://www.imim.es/comitesetics/en_index.html).
Provenance and peer review Not commissioned; externally peer reviewed.