Article Text

Abstract
Introduction: Lymphoedema-distichiasis syndrome (LD) (OMIM 153400) is a rare, primary lymphoedema of pubertal onset, associated with distichiasis. Causative mutations have now been described in FOXC2, a forkhead transcription factor gene. Numerous clinical associations have been reported with this condition, including congenital heart disease, ptosis, varicose veins, cleft palate, and spinal extradural cysts.
Subjects: We report clinical findings in 74 affected subjects from 18 families and six isolated cases. All of them were shown to have mutations in FOXC2 with the exception of one family who had two affected subjects with lymphoedema and distichiasis and linkage consistent with the 16q24 locus.
Results: The presence of lymphoedema was highly penetrant. Males had an earlier onset of lymphoedema and a significantly increased risk of complications. Lymphatic imaging confirmed the earlier suggestion that LD is associated with a normal or increased number of lymphatic vessels rather than the hypoplasia or aplasia seen in other forms of primary lymphoedema. Distichiasis was 94.2% penetrant, but not always symptomatic. Associated findings included ptosis (31%), congenital heart disease (6.8%), and cleft palate (4%). Other than distichiasis, the most commonly occurring anomaly was varicose veins of early onset (49%). This has not been previously reported and suggests a possible developmental role for FOXC2 in both venous and lymphatic systems. This is the first gene that has been implicated in the aetiology of varicose veins.
Conclusion: Unlike previous publications, the thorough clinical characterisation of our patients permits more accurate prediction of various phenotypic abnormalities likely to manifest in subjects with FOXC2 mutations.
- primary lymphoedema
- distichiasis
- FOXC2
- varicose veins
Statistics from Altmetric.com
Lymphoedema-distichiasis syndrome (LD) (OMIM 153400) is a rare, primary lymphoedema of pubertal onset, associated with distichiasis. Distichiasis is a congenital anomaly in which accessory eyelashes occur along the posterior border of the lid margins in the position of the Meibomian gland orifices. The accessory eyelashes may be represented by a few cilia or (less commonly) an additional regular, well formed row. Normally, the Meibomian glands act as modified sebaceous glands in which the ducts open directly onto the ocular surface to release sebum. The association of distichiasis of the lids with lymphoedema of the lower limbs was probably first described in 1899.1 Since that time, numerous clinical associations have been reported in this condition, including congenital heart disease, ptosis, varicose veins, cleft palate, and spinal extradural cysts.
The familial nature of primary lymphoedema in general and lymphoedema-distichiasis in particular has long been recognised.2–4 The gene for Milroy disease (congenital familial lymphoedema) has recently been mapped to chromosome 5q35.3 and probable causative mutations found in the VEGFR3 gene.5,6 Most families sharing this phenotype appear to be consistent with linkage to this region (A Evans, personal communication). The most common type of primary, familial lymphoedema is that first described by Meige (isolated pubertal onset lymphoedema),3 but to date no locus has been reported.
Recently, lymphoedema-distichiasis was mapped to chromosome 16q247 using three families with clear dominant inheritance of the condition. With the addition of further members from the largest family, the map distance was reduced to less than 2 Mb.8 A previously reported neonate with congenital lymphoedema was found to have a de novo balanced translocation involving chromosome 16 and the Y chromosome (t(Y;16)(q12;q24.3). Further studies showed that the chromosomal breakpoint was in the critical region for lymphoedema-distichiasis. The breakpoint did not appear to disrupt a gene, so candidate genes in the immediate region were considered. Two unrelated families with LD were found to have mutations in one of these genes, FOXC2. Subsequently, seven mutations were found in this gene in seven additional families with lymphoedema-distichiasis.9,10 A further 34 novel mutations in FOXC2, primarily small insertions and deletions, have now been characterised in our series of patients11 ( R Bell, personal communication). Another recent study looked at 86 families with primary lymphoedema and eleven of these families were shown to have a mutation in FOXC2. All but one of these families had distichiasis in one or more affected member.
In this paper we describe the clinical and molecular features of this condition as found in a group of 18 families and six isolated cases of lymphoedema-distichiasis syndrome.
METHODS
Eighteen familial and six isolated cases of distichiasis were ascertained from lymphoedema clinics (PSM and KGB), the operating lists of an ophthalmologist specialising in adnexal surgery (JROC), and from clinical geneticists in the UK. Probands were included if they had distichiasis and primary lymphoedema or if they had one of these features and a family history of the other feature. Informed consent was obtained from all participants and Ethics Committee approval obtained. Most probands and family members were examined looking specifically for signs of distichiasis, lymphoedema, and other known associations with LD. The authors examined all but six subjects. These six are included as reliable clinical information was available from hospital notes, photographs, and family members. In addition, all six had FOXC2 mutations. Blood or buccal samples were collected from all willing subjects and screened for mutations in the transcription factor gene FOXC2. The methods used for identifying the mutations have been previously described in detail.11 Families and sporadic cases were included in the study if a mutation in FOXC2 was identified or linkage to the gene confirmed (one family). There were a total of 74 affected subjects (43M:31F). Isotope lymphoscintigraphy was obtained on 16 subjects and 10 patients had previously had direct radiocontrast lymphangiography.
Penetrance for lymphoedema and difference in age of onset between males and females was calculated by excluding probands, to avoid ascertainment bias. Chi-squared and Cox regression analysis was used to assess statistical significance.
RESULTS
The mutations identified in our series of patients are listed in table 1. Most have previously been reported,11 but there are nine novel mutations. These are three point mutations, three single base insertions, two deletions (one 1 bp and one 8 bp deletion), and one 4 bp duplication. Two of the point mutations caused premature stop codons. The third was a missense mutation, and is only the second in FOXC2 so far identified as likely to give rise to LD, all the remainder being nonsense or frameshifts.9–12 As the G362A (R121H) change is found in an isolated case of LD, it is impossible to prove that this is the causative mutation in this person. However, this is a highly conserved amino acid, being one of only 11 that are identical in 24 members of the forkhead family in the 100 amino acid DNA binding domain.13 It is also adjacent to the less well conserved isoleucine that is mutated to methionine in Axenfeld-Rieger syndrome with glaucoma in FOXC1.14 Together with the fact that this mutation was not seen in 100 control chromosomes, this is compelling evidence that this missense mutation does produce LD in this person.
FOXC2 mutations found in a series of familial and isolated cases of lymphoedema-distichiasis. Those marked with an asterisk have previously been published11
One family showed no mutations but was consistent with linkage to the FOXC2 locus. There were two affected and three unaffected subjects. The two affected brothers both had bilateral lower limb lymphoedema, distichiasis, and ptosis. This pedigree generated a two point maximum lod score of 1.0 at 𝛉=0 for D16S520. As there has been no evidence for genetic heterogeneity in this condition, and both affected subjects had distichiasis and lymphoedema, this was felt to be highly suggestive of linkage to FOXC2.
Lymphoedema
Clinical aspects
In our series of 74 patients, 57 patients had clinical evidence of lymphoedema (36M:21F). Fourteen patients had no evidence of lymphoedema (9F:5M), two were thought probably to have lymphoedema, and one patient refused to be examined. Of the 14 with no evidence of lymphoedema (age range 1 to 34 years), six were under the age of 11 and therefore could yet develop lymphoedema.
The age of onset of the lymphoedema is shown by the cumulative risk graph (fig 1). There were 20 informative males and 22 informative females. Only three of the males were unaffected with lymphoedema at the time of the study, whereas eight women were unaffected. However, the age of onset in males is earlier and this may account for the difference. The earlier age of onset in males was statistically significant (p=0.015, using Cox regression analysis with one degree of freedom on SPSS version 10, Exp (B) = 2.628 with 95% confidence limits of 1.209 to 5.711). Half of the males are already affected by the age of 11 years, whereas only half the females are affected by their early 20s. The penetrance in this series appeared to be complete by the 40s, but there are very few informative older subjects.
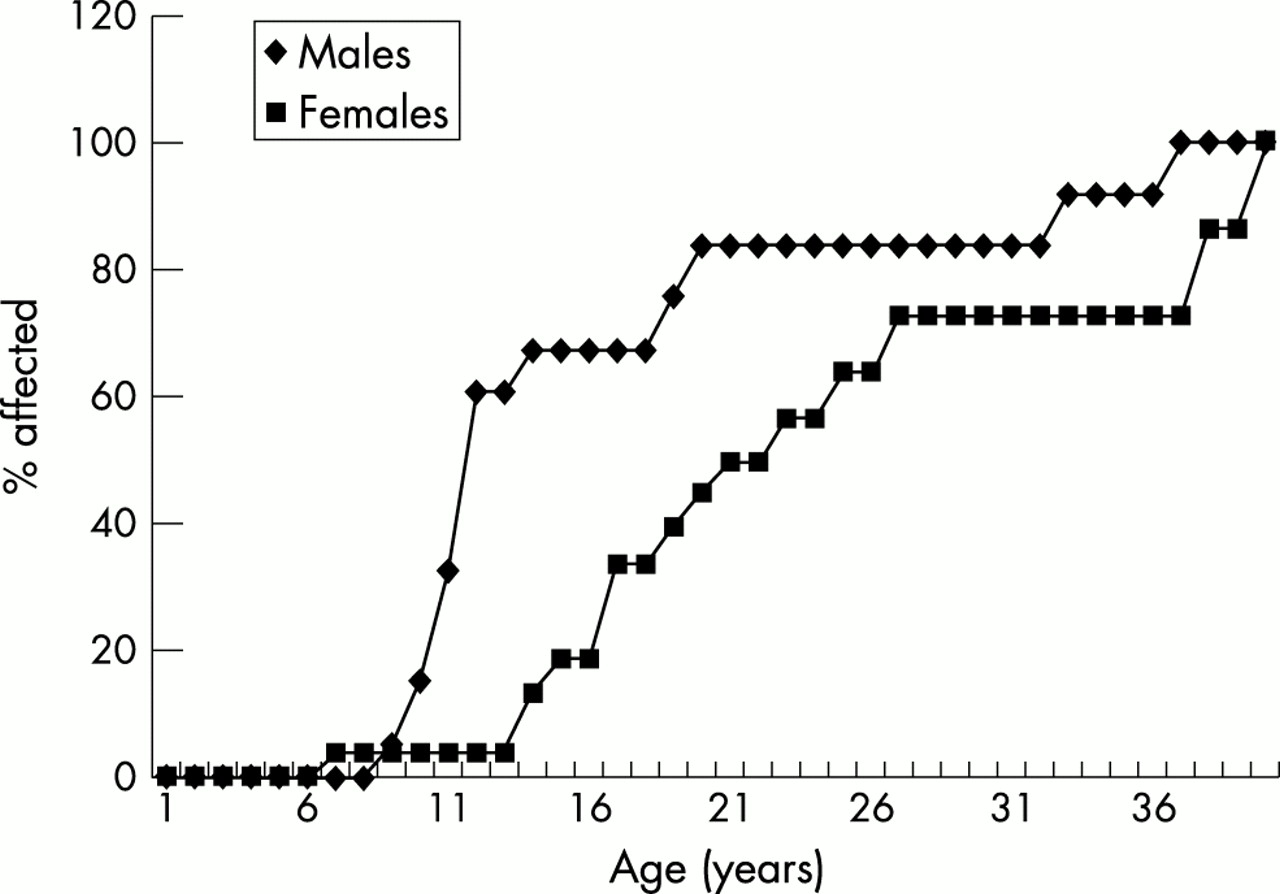
Cumulative penetrance of lymphoedema in our group of 74 cases.
The severity of the lymphoedema varied within and between families. Mild lymphoedema was defined as swelling confined to the lower leg, moderate lymphoedema to the knee, while marked lymphoedema was defined as swelling extending into the thigh. This information was not available for one affected female. Eleven percent (4/36) of males and 25% of females (5/20) had mild lymphoedema. There were 58.3% of males (21/36) and 60% of females (12/20) with moderate lymphoedema. There was a greater proportion of males with marked lymphoedema, 30.5% (11/36) compared with 15% of females (3/20), but this did not reach statistical significance (χ2, p=0.25).
The lymphoedema was usually bilateral and predominantly asymmetrical. In a minority (three patients) the lymphoedema was unilateral.
In most cases no precipitating factor was identified (37); however, in 27 (20M:7F) of these cases the age of onset was between 10 and 19 years (that is, may have been related to puberty). Precipitating factors included injury or infection (5) (such as insect bites, ingrowing toe nails, surgery) and in females the oral contraceptive pill and pregnancy (6).
Males were much more likely than females to develop cellulitis or infection in the oedematous leg. Where the information was available, 64.7% (22 of 34) of males and 25% (4 of 16) of females reported one or more episodes of infection. This was a statistically significant finding (χ2, p= 0.02).
Management of the lymphoedema included compression stockings, bandaging, and massage. The stockings provided some reduction in swelling and discomfort but were often uncomfortable in the summer months and cosmetically unacceptable to many patients. Two patients underwent bulk reducing surgery which was unsuccessful.
Isotope lymphoscintigraphy and lymphangiography
Lymphoscintigram results were consistent with lymphoedema in nine of 11 subjects tested, with abnormally low uptake of radioactive colloid in ilioinguinal nodes at both 30 minutes and one hour. Two females, aged 23 and 24, showed normal uptake at both 30 and 60 minutes and no clinical evidence of lymphoedema; however both were found to have asymptomatic distichiasis. In some cases lymphatic function was within normal limits in terms of nodal uptake, but images showed increased lymph conducting pathways and dermal backflow indicating lymph reflux (figs 2 and 3).

Normal lymphoscintigram.

Lymphoscintigram of patient with LD showing reflux of tracer back down into the lower leg.
Lymphangiograms, now rarely performed, had previously been performed on 10 subjects. Five of these were reviewed and showed increased nodal tissue with small multiple nodes extending into the mesentery. The number of lymph channels was at the upper limit of normal (figs 4 and 5). One of these lymphangiograms has been previously reported.15

Lymphangiography in an affected subject showing increased nodal tissue with multiple small lymph nodes in the mesentery.
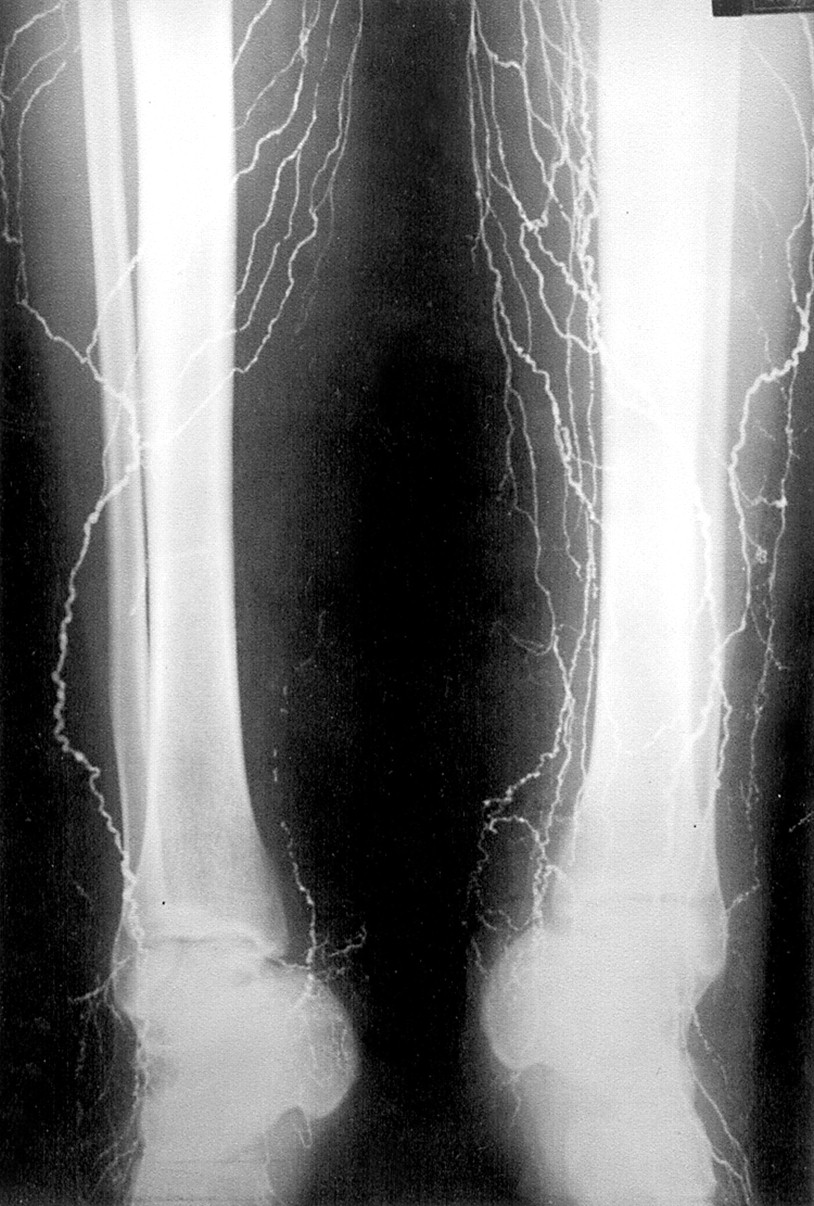
Lymphangiography in an affected subject showing that the number of lymphatic channels are at the upper limit of normal.
Distichiasis
Distichiasis (fig 6) was present in 38 males (92.7%) and 28 females (96.6%) (overall prevalence of distichiasis was 94.3%). It was not present in four patients (3M:1F) and two patients were not examined for distichiasis. In two further patients, the findings were uncertain (there were one or two accessory eyelashes in the inner canthus only). The distichiasis was not always diagnosed at birth but usually during childhood or puberty. However, it is felt that the distichiasis is probably present at or shortly after birth but frequently not diagnosed until later. Six affected patients had no symptoms of distichiasis and were only discovered on clinical examination during this study. The distichiasis was usually confined to a few sparse, fine eyelashes. They occurred on the inferior and superior eyelid and were more often central and lateral than medial. In one male carrier of a FOXC2 mutation, there was no distichiasis but the Meibomian glands appeared prominent.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Congenital distichiasis. This was asymptomatic and has not required treatment despite the aberrant lashes resting on the cornea.
Complications occurred in 73.7% (45 of 61) of the patients in whom information was available. These included mild corneal irritation, photophobia, recurrent conjunctivitis, and styes. There was no significant difference in the incidence of complications between males and females. The distichiasis was managed in a number of different ways including epilation (plucking), cryotherapy, electrolysis, lid splitting operations, and laser treatment. Most of these measures gave only temporary relief.
Varicose veins
Varicose veins are common in the general population, increasing in prevalence with age. A study in 1978 showed a prevalence of 10% in the population between the ages of 25 and 34 years rising to 50% over the age of 64.16 Previously, varicose veins have been shown to be more common in patients with lymphoedema, with a prevalence of 25%,17 although the type of lymphoedema was not specified. In one large lymphoedema-distichiasis family, linked to chromosome 16q24, all subjects affected with lymphoedema also had abnormal findings when venous function was tested using light reflective rheography.18 The findings on doppler ultrasound of the veins in a 19 year old, of bilateral incompetence at the saphenofemoral junction and incompetent mid-calf perforator veins plus moderate reflux in the deep veins, suggested congenital abnormality of the deep and superficial veins.
In our group of subjects, varicose veins were notable owing to both early onset and increased prevalence compared with the general population. Forty-nine percent (33 of 67) of subjects in this study were found to have varicose veins with age of onset between 7 and 28 years. Males and females were found to be affected equally often and the varicose veins were frequently present before the onset of lymphoedema.
Ptosis
Congenital ptosis is not an uncommon finding in the general population. However, it was noted in 31% (14M:9F) of patients with LD. Severity ranged from mild asymptomatic cases to extreme bilateral cases requiring early surgery to allow correct development of the visual pathway. Sixty percent (14 of 23) of cases of ptosis were bilateral but most were mild. Six of the 23 (26%) affected patients had marked ptosis, all of whom required surgery (three bilateral, three unilateral).
Congenital heart disease
Congenital heart disease occurs in the white population at the rate of approximately 1%.18 In this series, the rate was considerably higher at 6.8% (five cases). There were two patients with Fallot's tetralogy, one ventricular septal defect, one pulmonary defect (not specified), and one patent ductus arteriosus. Cardiac arrhythmias have been reported in association with this condition.20 Four of these patients have been investigated in the past for palpitations, and two from the same family have had episodes of sinus bradycardia. None of these patients required treatment.
Cleft palate
Cleft lip and/or palate occurs in the European population at a rate of 7.9 per 10 000 live births.21 In this study group, three males (no females) had a cleft palate (4%). Each of these occurred in isolation (that is, not within the same family). One of these had Pierre-Robin sequence, one had a cleft of the hard palate, and another soft palate involvement only.
In one family, the father required a tracheostomy in infancy for a “throat blockage”. His medical records were not available but it is possible that he had Pierre-Robin sequence. His son was reported as having a subglottic stenosis.
Other associations
Scoliosis was reported in two patients. One previously reported patient had scoliosis in association with rib fusion, neck webbing, double uterus, and bilateral, severe ptosis.22 Only one other subject had neck webbing.
Five patients reported renal abnormalities. Two had nephritis in early childhood, one had a duplex kidney, one had multiple urinary tract infections, and one patient required a renal transplant for recurrent pyelonephritis.
There were no reports of spinal extradural cysts, but there was one spinal tumour (fibrillary astrocytoma).
There was only one affected subject who had a hydrocele at birth. An affected female had an ovarian varicocele.
Three patients had strabismus, one requiring an operation at the age of 2 years. Other eye abnormalities included early cataracts (n=1) and corneal dystrophy (n=1).
An affected 4 year old boy with very marked distichiasis also had moderate learning difficulties and autistic features. One of the affected males had a daughter with learning difficulties but examination and testing were refused. There were no dysmorphic facies associated with this condition, but synophrys was noted in a number of cases.
DISCUSSION
Clinical practice
Lymphoedema-distichiasis is said to be a very rare cause of primary lymphoedema. However, as is clear in this report, 18 families and six isolated cases have been identified in the United Kingdom. There are very few publications on this condition and the phenotype is not well characterised. Many cases of LD have possibly been labelled as Meige disease as the patients have not been asked about or examined for distichiasis. The lymphoscintigrams may give an indication that this is the underlying condition. In LD they characteristically show initial uptake of tracer from the feet to the groin nodes with subsequent reflux of tracer back down into the lower leg (fig 3), whereas patients with Milroy and Meige syndromes simply show lack of uptake of the isotope and little or no node uptake. Lymphangiography, which is rarely performed now, shows that the number of lymphatic vessels is at the upper limit of normal (compared with the hypoplasia found in Meige syndrome) and multiple small lymph nodes in the mesentery. Therefore, although both may present with pubertal lymphoedema, the underlying mechanism appears to be very different. The cause of the lymphatic malfunction in the presence of normal or increased numbers of lymph vessels is not clear. It is likely that these lymphatic vessels function poorly or they may be the result of a proximal abnormality or obstruction.
The lymphoedema in LD usually appears during puberty or later (occasionally it presents in childhood). Results here prove, for the first time, that it is possible to develop lymphoedema of genetic origin well into adult life. This has been suspected but there has been no evidence to support the hypothesis. Unlike Meige syndrome, which has an increased incidence and severity in females, LD affects males at an earlier age and possibly more severely. Infections, such as cellulitis, seem to cause an irreversible exacerbation of the swelling and are more frequent in males. There may be hormonal factors involved which are not yet understood. This is suggested by the fact that most subjects develop lymphoedema around the time of puberty and females report onset associated with the oral contraceptive pill and pregnancy.
Distichiasis is highly penetrant. It is present from an early age and probably at birth. It is often the first indicator that a person is affected. Frequently, distichiasis is associated with corneal irritation, photophobia, and conjunctivitis. Occasionally it is asymptomatic, usually because the lashes curl away from the cornea, but in some cases this may be the result of corneal hypoaesthesia.23 The problems associated with the distichiasis are, therefore, unrelated to the number of aberrant eyelashes present. The extra eyelashes can often be seen on close inspection by the naked eye, but in some cases slit lamp examination is required. It has been proposed by Fox24 that distichiasis occurs separately from lymphoedema. His report looked at 78 patients with a diagnosis of distichiasis; however, there is no mention of lymphoedema. In our series, many ascertained from an adnexal surgeon, there were no families in which lymphoedema was absent, except in those cases where the only affected members were very young and in whom the lymphoedema may not yet have developed.
There was a very high frequency of varicose veins of early onset and often with no predisposing factors. Lymphatic vessels and veins have the same embryological origin, so FOXC2 probably has a role in the development of both.25 This is the first gene that has been implicated in the aetiology of chronic venous disease. It is likely that the abnormality in the veins aggravates the lymphoedema by increased capillary filtration owing to the venous hypertension.
Interestingly, there were no cases of spinal extradural cysts (despite a number of published reports); however, these could be asymptomatic, as noted by Schwartz et al.26 Recently, a FOXC2 mutation has been described in a family with dominant inheritance of cleft palate and distichiasis.27 Cleft palate occurred in three of our patients from different families. All three had novel FOXC2 mutations and were in different domains to the mutation described by Bahau et al.27
Ptosis occurred frequently in this group of patients. This suggests another developmental role for FOXC2. Interestingly, blepharophimosis, ptosis, epicanthus inversus syndrome (BPES) has been shown to be caused by mutations in another forkhead transcription factor gene, FOXL2.13
There were a number of unusual clinical features in this series of patients. Many of these are likely to be coincidental findings. Renal abnormalities and learning difficulties have not been previously reported and may not be related to the lymphoedema-distichiasis syndrome.
Genetics
The gene for this condition, FOXC2, formerly known as MFH-1, is a forkhead transcription factor gene encoding a 2.2 kb transcript with a single exon coding region which is highly GC rich. It has not previously been linked to any human disease. However, another forkhead transcription factor gene, FOXC1 (FKHL-7), which is highly homologous to FOXC2, has been reported as causing anomalies of the anterior segment of the eye, which are dominantly inherited.14
In families with FOXC2 mutations, there appears to be no clear genotype-phenotype correlation. There was much variation in expression of the disease even within families in this series. This is consistent with a model of haploinsufficiency, with the intra- and interfamilial variation most likely the result of stochastic effects or interaction with other genes in the FOXC2 pathway.
Genetic heterogeneity has been postulated in a single family with no identified mutation of FOXC2.28 However, no linkage data were available and three members of the family had keratoconus (two required corneal transplantation), a feature not seen in this series of 74 patients. From our series we have been unable to show heterogeneity. In one family, with two subjects clearly affected with both distichiasis and lymphoedema, a FOXC2 mutation was not found. In this family, linkage data supported linkage to the FOXC2 locus. Further analysis will be required as our analyses so far would not exclude large deletions or rearrangements or mutations in the promoter or 3` untranslated region.
FOXC2 has recently been proposed to be important in the prevention of obesity and diet induced insulin resistance.29 It is postulated that an increase in the expression of FOXC2 leads to a lean and insulin sensitive phenotype and, conversely, a decrease, as would be expected in the haploinsufficiency model, should lead to obesity. Although in our series body mass index was not recorded, there was no obvious excess of obese subjects or diabetics.
Understanding development
LD is an uncommon cause of primary lymphoedema but probably not as rare as previously considered. The distichiasis is a highly penetrant feature and an important indicator of this disease. Much of the genetics of primary lymphoedema of pubertal onset remains unknown, presumably because this group has genetic heterogeneity. The distichiasis in this condition has been a distinguishing feature identifying a particular group of primary lymphoedema and therefore leading to identification of the causative gene.
The aspects of the phenotype shown here are of interest when considering the possible developmental roles of FOXC2 in human development. This is especially true of the venous insufficiency associated with LD, as it suggests that the gene plays an important role in the genesis of both venous cardiovascular and lymphatic systems. A recent publication supports this.30
The lymphoedema, in itself, is not life threatening but is the cause of much morbidity. It is difficult to manage and many of the more severely affected patients suffer from its cosmetic appearance. Understanding the function of this gene, and its developmental pathway, may help in identifying other important genes involved in the development of the lymphatic system.
Acknowledgments
RB and GB were supported by the British Heart Foundation. AHC wishes to acknowledge the support of the Bluff Field Charitable Trust. We wish to thank The Birth Defects Foundation for funding, Dr David Goudie, Consultant Clinical Geneticist, Ninewells Hospital and Medical School, Dundee for referral of patients, and John Simpson for statistical advice.