Article Text

Abstract
Background: Cross sectional studies have shown that 1-2% of patients with neurofibromatosis 1 (NF1) develop malignant peripheral nerve sheath tumours (MPNST). However, no population based longitudinal studies have assessed lifetime risk.
Methods: NF1 patients with MPNST were ascertained from two sources for our north west England population of 4.1 million in the 13 year period 1984-1996: the North West Regional NF1 Register and review of notes of patients with MPNST in the North West Regional Cancer Registry.
Results: Twenty-one NF1 patients developed MPNST, equivalent to an annual incidence of 1.6 per 1000 and a lifetime risk of 8-13%. There were 37 patients with sporadic MPNST. The median age at diagnosis of MPNST in NF1 patients was 26 years, compared to 62 years in patients with sporadic MPNST (p<0.001). In Kaplan-Meier analyses, the five year survival from diagnosis was 21% for NF1 patients with MPNST, compared to 42% for sporadic cases of MPNST (p=0.09). One NF1 patient developed two separate MPNST in the radiation field of a previous optic glioma.
Conclusion: The lifetime risk of MPNST in NF1 is much higher than previously estimated and warrants careful surveillance and a low threshold for investigation.
- neurofibromatosis 1
- malignant peripheral nerve sheath tumours
- NF1, neurofibromatosis 1
- MPNST, malignant peripheral nerve sheath tumours
Statistics from Altmetric.com
NF1 is an autosomal dominant condition with a birth incidence of about 1 in 2500 and a prevalence of 1 in 4000.1 Most studies of NF1 are biased towards childhood disease and little is known of the natural history of the disease in adulthood. Nonetheless, recent longitudinal studies2,3 and studies of death certification4,5 have shown that a large proportion of NF1 patients succumb to malignancy in mid-adult life. Studies from our own centre have shown that MPNST is often misdiagnosed and careful review of histology is often necessary.6 Previous studies have shown that 20-50% of patients with MPNST also have NF1.2,7 However, cross sectional studies of NF1 show that only 1-2% of NF1 patients have MPNST.1 Studies based on specialist hospital series have shown much higher rates,8 but these studies are inevitably subject to ascertainment bias. In order to assess the true risk of MPNST in NF1, we have used two sources of information (The North West Regional Cancer Register and NF1 Genetics Register) in an effort to obtain all coexistent patients over a 13 year period.
METHODS
The North West Regional Cancer Registry covers a region of north west England, based around Manchester, with a population of 4.1 million. The Cancer Registry ascertains patients with malignancies of all sites, as well as benign central nervous system tumours, from pathology records and death certificates. We reviewed the Cancer Registry for patients with MPNST (ICD-0: M9540/3 and 9560/3). There is very high overall coverage, although misclassification of MPNST does occur at peripheral hospitals.6 In order to obtain all possible patients with coexistent NF1 and MPNST, we also reviewed our North West Regional NF1 Genetic Register. This covers the same region and proactively obtains details of NF1 from families with the condition. Currently, the NF1 Register has details of 730 patients with NF1. The study period was 1984-1996 inclusive. We reviewed hospital notes for all patients with MPNST who were identified for the study. To confirm a diagnosis of NF1, histology reports and details of other typical NF1 disease features were reviewed (for example, café au lait macules and neurofibromas with respect to the US National Institutes of Health diagnostic criteria9). Histology from all sporadic MPNST patients was reviewed to avoid misdiagnosis.
Death details from the registries were used to calculate survival rates and death certificates were reviewed to determine cause of death. Vital status was determined as of 1 April 2001. Five year survival was determined using Kaplan-Meier curves. NF1 MPNST cases who were identified on the periphery of the region during the study period were included for the survival analysis only. The Mann-Whitney U test and Wilcoxon (Gehan) statistic were used to test between group differences in age at diagnosis and survival (fig 1).

Kaplan-Meier analysis of survival for NF1 MPNST patients and people with sporadic MPNST (Wilcoxon (Gehan) statistic, p=0.09).
RESULTS
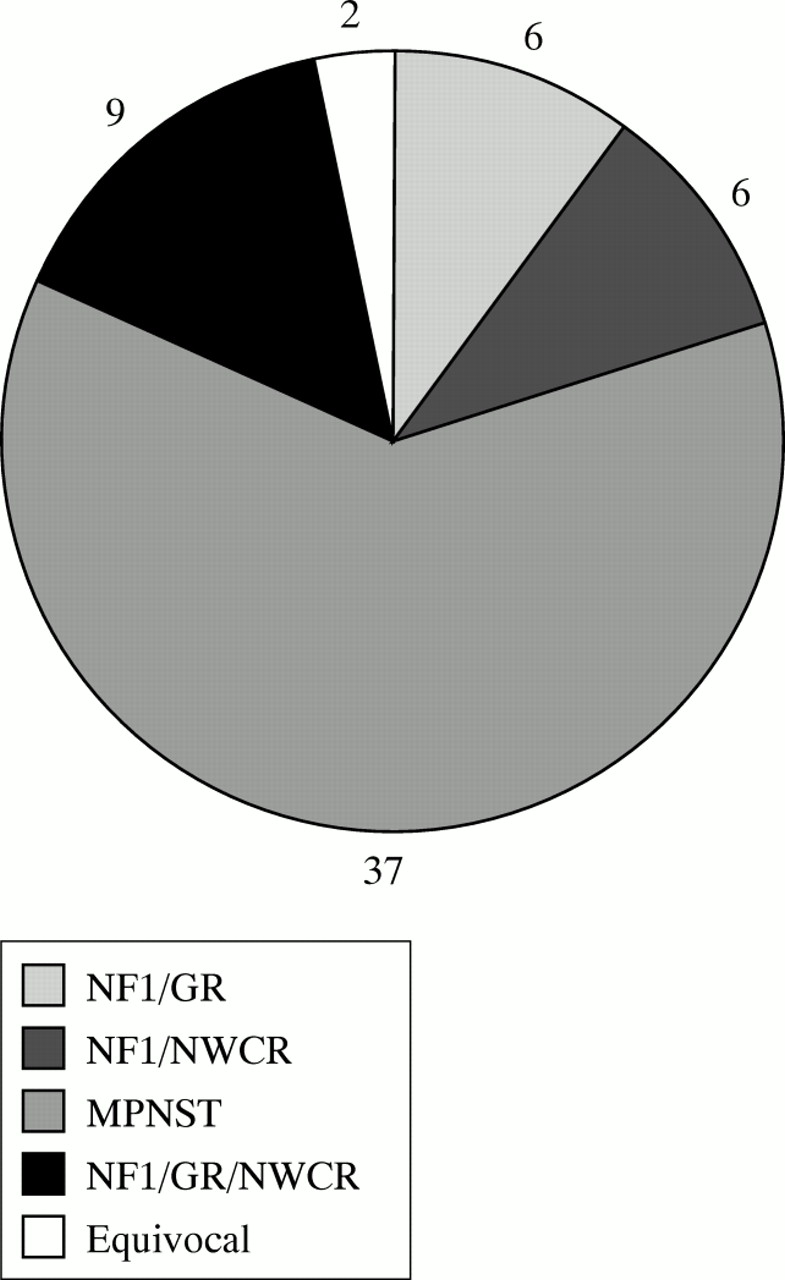
The Cancer Registry contained 74 entries of MPNST for the study period. Fifty-four patients were confirmed from hospital notes and pathology records as having MPNST. Six patients were excluded as having different diagnoses, including four with benign schwannoma. The notes of the remaining 14 patients were unavailable for review. After careful review of hospital notes, 15 of the 54 MPNST patients were confirmed as having NF1. Two patients were equivocal, one having two neurofibromas in the area around the MPNST but no other features, and another having insufficient information to confirm or exclude NF1. Nine of 15 NF1 MPNST patients on the Cancer Registry were also in the NF1 Register (their diagnosis being confirmed clinically), which identified nine additional patients with MPNST, for a total of 24 patients with NF1 and MPNST over the 13 year period. Three of the nine patients were registered as MPNST in the Cancer Registry, but were not detected on the initial search; three were just outside the regional boundaries (one recorded in the Cancer Registry) and the remaining three had other disease classifications on the Cancer Registry (liposarcoma, schwannoma, or carcinomatosis). However, histological review confirmed MPNST (total numbers for strict region and time period are shown in fig 2). The median age at diagnosis in NF1 MPNST was 26 years (range 16-77 years), compared to 62 years (range 19-89 years) in patients with sporadic MPNST (p<0.001). Two NF1 patients had previous cranial irradiation for optic glioma. One of these (patient 11, table 1) developed a right temporal MPNST (which was completely excised) 24 years after irradiation for a left optic glioma and a left submandibular MPNST three years later. The other patient (patient 2) developed a brachial plexus MPNST 18 years after irradiation. Eight additional patients with MPNST were identified from the NF1 Register, either outside the study period or outside the strict geographical boundaries of the regional genetics catchment area. These patients were not used in the survival analysis or in the frequency calculations. One of these patients (patient 5) developed a MPNST 17 years after radiotherapy for optic glioma.
Characteristics of patients with MPNST
{kind=link}
{kind=link}
Breakdown of the 60 confirmed cases of MPNST in the strict North West Regional area over the 13 year period.
Location
The locations of all tumours identified from both the Cancer Registry and the NF1 Register are shown in table 1. Twelve of 32 tumours (38%) occurred in deep locations with no superficial signs of a plexiform tumour. Many others occurred on major nerve routes such as the brachial plexus or sciatic nerve, but it is not clear whether these all grew from existing nodular tumours. Some tumours did occur in definite existing plexiform tumours. Few tumours occurred at extremes of the limbs that would allow radical surgery such as amputation. All tumours presented with pain or rapid growth, apart from patient 28, who had dysphonia and dysphagia.
Survival
Fig 1 compares the survival from diagnosis of NF1 patients with MPNST with that of people with sporadic MPNST. The five year survival rate was 42% for the 37 sporadic MPNST patients (95% confidence interval 26-58%), compared to 21% for the 24 NF1 MPNST patients (95% confidence interval 5-37%) (median survival 2.8 years and 1.3 years, respectively, p=0.09). In those who were diagnosed at less than 60 years of age (22 NF1 MPNST patients and 16 sporadic MPNST patients), the five year survival rate was 50% for sporadic MPNST patients, but only 18% for those with NF1 MPNST (median survival 7.0 years and 1.0 year, respectively, p=0.09). In sporadic MPNST patients, the five year survival rate was 32% in males and 56% in females (median survival 1.7 years and 6.7 years, respectively, p=0.13).
NF1 MPNST lifetime risk
Assuming a population prevalence of 1 in 4000 for NF1,1 the annual incidence of MPNST in NF1 patients is 1.6 per 1000 (only strict regional patients included, 21/13). If NF1 patients live to 75-80 years, the lifetime risk is 12-13%, respectively. However, the birth incidence of NF1 may be as high as 1 in 2500,1,9 which corresponds to an annual incidence of 1.0 per 1000 and a lifetime risk of 8%. This is in accord with our previous estimates of lifetime risks based on actuarial analysis of our NF1 Register,10 in which there are now 32 known cases of MPNST among 730 NF1 patients. If life expectancy is lower than 75-80 years (for example, 65 years5), prevalence decreases and annual incidence increases, yielding a lifetime risk of about 10%.
DISCUSSION
The association between MPNST and NF1 is well known.7 Initial reports that were based on specialist series found high risks of MPNST in NF1,8 but more recent population based series have tended to downplay the overall risk.7 However, most of these studies have inherent biases; specialist series will not have many mildly affected patients with NF1, and attempts to obtain 100% ascertainment of NF1 patients in an area can initially only examine cross sectional estimates of risk.1,11
More recent long term longitudinal studies of NF1 patient cohorts are more likely to arrive at an accurate estimate of risk.2,3 However, neither of two recent large studies calculated lifetime risk. Nonetheless, a study of 1475 patients in North America3 found 34 new cases of MPNST over a 20 year period. This equated to a risk of 1.3-6.8 per 1000 from the first to the fifth decades of life. The number of older patients was small; the median age of NF1 patients without MPNST at initial assessment was only 14.7 years. If extrapolated, this corresponds to a lifetime risk of as much as 20-24% (using an average annual risk of 3 per 1000). The main drawbacks of this study were the lack of older patients and the probable ascertainment bias towards more severely affected NF1 patients.
Our study is entirely population based for MPNST and it is likely that there is near total ascertainment for NF1 related MPNST using our dual approach of ascertainment through the NF1 Register and the Cancer Registry. Sporadic MPNSTs could be underestimated owing to the possible misclassification of other sarcomas.6 While the calculation of lifetime risk involves a number of assumptions about the incidence and prevalence of NF1 and life expectancy, our previous actuarial analysis in our NF1 Register resulted in a similar lifetime risk of 10%.10 This was true in spite of the fact that patients were excluded if they were ascertained only from the Cancer Registry. In contrast to the large North American study,3 we did not find an even age distribution of NF1 related MPNST; there was a definite peak in the mid-30s. This contrasts with sporadic MPNST, in which the risk is highest after 60 years of age. It is likely that our sporadic MPNSTs are more representative of a population series than most previous studies, since elderly patients may not have been referred to specialist centres. This probably explains the much older median age for our patients than in previous studies.12–14 Not surprisingly, NF1 patients were significantly younger than sporadic patients. Twenty-eight per cent of the Cancer Registry MPNST patients in whom assessment was possible had NF1, in keeping with previous studies.7,8
A number of previous reports have highlighted the poorer prognosis in NF1 related MPNST compared to sporadic tumours.15–17 It is likely that this is because of later presentation with the malignant tumour or false reassurance by medical staff. A patient without NF1 is likely to be perturbed by a new, rapidly growing lump, particularly if it is painful. As patients with NF1 can have many lumps, they may become complacent about any new ones. Indeed, this may be compounded by attempts to downplay the risks of MPNST in NF1. While we cannot be certain that this is the cause of the poorer outcome (it could be the result of other NF1 specific features of the tumour), it is clear that radiotherapy and chemotherapy are of little value and only complete surgical excision before metastasis is likely to give a good prognosis.8,12,13,17 The severity of other malignant lesions, like optic gliomas, which have a better prognosis in NF118,19 than in sporadic patients would also support late presentation as the main reason for poor outcome.
The NF1 patient support group publications tend to quote a lifetime risk of MPNST of 1-2%. In addition to correcting this to a higher lifetime risk of NF1 related MPNST as indicated by the results of this study, NF1 patients need to be aware that MPNST may not occur in a pre-existing plexiform tumour, but from a deeper location. Most of the NF1 patients in our series developed tumours in deep seated locations and only a small proportion occurred in typical cutaneous plexiform tumours. This adds to the findings in the North American study,3 where 36% of tumours arose in deep areas with no previous plexiform tumour. We found a slight trend to a worse prognosis in males with sporadic MPNST, which accords with previous observations.17
The risk of radiation induced MPNST has been largely overlooked in recent years. However, there are several published reports relating to patients with and without NF1.7,12,17 Ducatman et al12 described 12 patients with post-irradiation MPNST, of whom seven had NF1 with two having had radiotherapy for optic gliomas five and 17 years previously. More recently, Loree et al17 described two of four NF1 patients who developed MPNST after head and neck irradiation. We therefore concur with the previous reports in that radiotherapy should only be used when absolutely necessary in NF1,12,17 which is now probably not the case for optic glioma.18
We can make a number of recommendations based on the results of our study. (1) Patients with NF1 should be made aware of the relatively high risk of MPNST with the likely symptoms of pain and rapid growth, and they should have easy access to expert opinion should these symptoms occur. (2) Better diagnostic tools such as PET scanning20 should be used in suspicious lesions, and prospective studies of PET scanning in the highest risk patients should be considered. (3) Radiation treatment in NF1 should only be used if there is clear clinical need and no equally effective alternative.
REFERENCES
Supplementary materials
Malignant peripheral nerve sheath tumours in neurofibromatosis 1
D G R Evans, M E Baser, J McGaughran, S Sharif, E Howard, and A MoranAuthors' Correction
The Kaplan Meier curve published in the article was an analysis from birth to current age or death rather than from diagnosis. The p value attached to the curve relates to the analysis from diagnosis. The correct figure of survival from diagnosis is provided here.Figure 1 Kaplan-Meier analysis of survival for NF1 MPNST patients and people with sporadic MPNST (Wilcoxon (Gehan) statistic, p=0.09).
[View Corrected Figure] (PowerPoint slide)The error is much regretted
Linked Articles
- Correction