Article Text

Abstract
Recent studies have shown that cryptic unbalanced subtelomeric rearrangements contribute to a significant proportion of idiopathic syndromic mental retardation cases. Using a fluorescent genotyping based strategy, we found a 10% rate of cryptic subtelomeric rearrangements in a large series of 150 probands with severe idiopathic syndromic mental retardation and normal RHG-GTG banded karyotype. Fourteen children were found to carry deletions or duplications of one or more chromosome telomeres and two children had uniparental disomy. This study clearly shows that fluorescent genotyping is a sensitive and cost effective method that not only detects cryptic subtelomeric rearrangements but also provides a unique opportunity to detect uniparental disomies. We suggest giving consideration to systematic examination of subtelomeric regions in the diagnostic work up of patients with unexplained syndromic mental retardation.
- mental retardation
- subtelomeric rearrangements
- uniparental disomies
- fluorescent genotyping
- MR, mental retardation
- CGH, comparative genomic hybridisation
- FISH, fluorescence in situ hybridisation
- UPD, uniparental disomy
- PFGE, pulse field gel electrophoresis
- DOP-PCR, degenerate oligonucleotide primed polymerase chain reaction
Statistics from Altmetric.com
- MR, mental retardation
- CGH, comparative genomic hybridisation
- FISH, fluorescence in situ hybridisation
- UPD, uniparental disomy
- PFGE, pulse field gel electrophoresis
- DOP-PCR, degenerate oligonucleotide primed polymerase chain reaction
Mental retardation (MR), defined as an intelligence quotient (IQ) below 70, is the most frequent serious handicap in children and young adults.1 Moderate to severe MR (IQ<50) affects 1% of the population and this prevalence increases to 2-3% if mild MR is included (50<IQ<70).2 MR can result from multiple causes including environmental factors, chromosomal anomalies, and monogenic disorders. Despite recent advances in cytogenetics and molecular genetics, the cause of the mental handicap remains unexplained in about 40% of cases.3
Taken together, chromosomal anomalies are believed to account for 4-28% of cases of MR.4 Because no practical way of screening the entire genome is available at present, efforts have first focused on rearrangements involving subtelomeric regions. Indeed, cryptic subtelomeric rearrangements undetectable using conventional cytogenetic methods have been observed in the α thalassaemia/MR syndrome (16pter),5, 6 Wolf-Hirschhorn syndrome (4pter),7 Miller-Dieker syndrome (17pter),8 and the cri du chat syndrome (5pter).9 Moreover, cryptic subtelomeric rearrangements have been found to account for 5-7.4% of moderately or severely mentally handicapped children.10–12
Several methods aimed at investigating telomere integrity have recently been devised. Fluorescence in situ hybridisation (FISH) uses telomeric probes directly to assess loss or gain of telomeric sequences.13 This method is a powerful diagnostic tool, but it remains costly and technically complex. Comparative genomic hybridisation (CGH) to metaphase chromosomes is also a valuable technique,14 but it can only detect deletions larger than 5 Mb and requires specific microscope and computer facilities. To overcome these limitations, we have recently developed a novel strategy based upon automated fluorescent genotyping to search for non-mendelian segregation of telomeric microsatellites.15
Here, we report on 14 subtelomeric rearrangements and two uniparental disomies in a series of 150 mentally retarded children (10.7%). We conclude that genotyping is an effective approach to detecting cryptic subtelomeric anomalies in children with severe MR and that idiopathic syndromic MR can result from uniparental disomy (UPD) of subtelomeric regions. We suggest that patients with unexplained syndromic MR should be systematically tested for subtelomeric rearrangements.
METHODS
Patients
A total of 150 children (79 boys and 70 girls, belonging to 125 families) born to unrelated parents and presenting with moderate to severe idiopathic MR (IQ below 50) were recruited from the Department of Genetics of the Hôpital Necker-Enfants Malades, Paris. The children were included in this study on the basis of the presence of at least one of the following additional criteria: (1) family history of MR, (2) overgrowth or failure to thrive, (3) behavioural problems (hyperactivity, aggression, or self-mutilation), (4) seizures, and (5) facial dysmorphism (fig 1) or clinical or radiological evidence of brain, trunk, or limb anomalies. Fig 2 summarises the frequency of each clinical feature associated with MR. In addition, all patients had a karyotype interpreted as normal using both RHG and GTG banding analysis at ISCN 400-500. The results of routine biochemical tests and haematological examinations were normal.

Facial appearance of children with de novo telomeric rearrangements.

Frequency of clinical features in children carrying telomeric rearrangements (black bars, n=16) and in the entire cohort (white bars, n=150).
Genotyping analysis
Blood samples were obtained from the probands and their parents and genomic DNA was isolated from EDTA anticoagulated blood by a salting out procedure. Fluorescent genotyping was performed as previously described.16
Chromosome and FISH studies
Metaphase spreads were prepared from phytohaemagglutinin stimulated blood lymphocyte cultures using standard procedures of hypotonic treatment and methanol/acetic acid fixation (3:1). RHG and GTG banding methods were performed according to standard protocols. Thymidine synchronisation and FUdR incorporation were used to obtain high resolution R and G banding.
Two sets of subtelomeric FISH probes were used for hybridisation. The first set of probes corresponds to the Chromoprobe Multiprobe T System commercially distributed by Cytocell and is composed of telomeric specific cosmid and PAC clones previously tested on unrelated subjects to exclude polymorphisms. The second set of probes is composed of 41 well characterised CEPH YACs specific for each subtelomeric region and located about 2-3 Mb away from the telomere. They were kindly provided by Thomas Haaf. YAC DNA was isolated by pulse field gel electrophoresis (PFGE) and amplified using the degenerate oligonucleotide primed polymerase chain reaction (DOP-PCR) procedure as previously described. Probes were labelled with biotin-16-dUTP or digoxigenin-11-dUTP (Boehringer-Mannheim) using a commercially available random priming kit (Gibco-BRL). Biotin labelled probes were detected using Texas Red (TR) conjugated to avidin and digoxigenin labelled probes were detected using fluorescein isothiocyanate (FITC) conjugated to anti-digoxigenin. Slides were counterstained with 4`,6`-diamidino-2-phenylindole (DAPI). Image capture and analyses were performed using a Zeiss Axiophot epifluorescence microscope equipped with the appropriate filter combination for detecting TR, FITC, and DAPI. The images were captured by a cooled CCD camera controlled using an image analysis system (Vysis). Ten hybridised metaphases were analysed for each probe.
RESULTS
Large scale systematic screening
A total of 150 mentally retarded children and their parents were analysed for segregation of telomeric markers. For each marker, the genotype of the child was determined and compared to the parental genotypes so as to detect (1) missing alleles (deletion), (2) the presence of a third allele (duplication), or (3) the presence of one or two alleles from one parent with no contribution from the other parent (uniparental isodisomy or heterodisomy respectively). Whenever the child was heterozygous for two different alleles identical to those of his parents, the genotype was regarded as normal. In case of uninformative polymorphism (that is, when the child was homozygous for one allele shared by both parents), the segregation of the closest microsatellite marker on the Genethon map was tested.
A total of 22 344 genotypes were determined and 46 cases of abnormal allele inheritance were reproducibly detected. In 22/150 cases, the child was heterozygous and carried an allele which was not present in his parents. Since non-paternity was excluded in all cases, these patterns were more likely the result of microsatellite instability in one of the parental alleles, so that the final repeat length was different. In 24/150 cases, absence of one of the parental alleles was suggestive of a deletion. Finally, we found two cases of abnormal segregation of the telomeric markers of chromosomes 17q or Xq, suggestive of a uniparental disomy with two alleles originating from the same parent and no contribution of the second parent.
Refined characterisation of telomeric anomalies
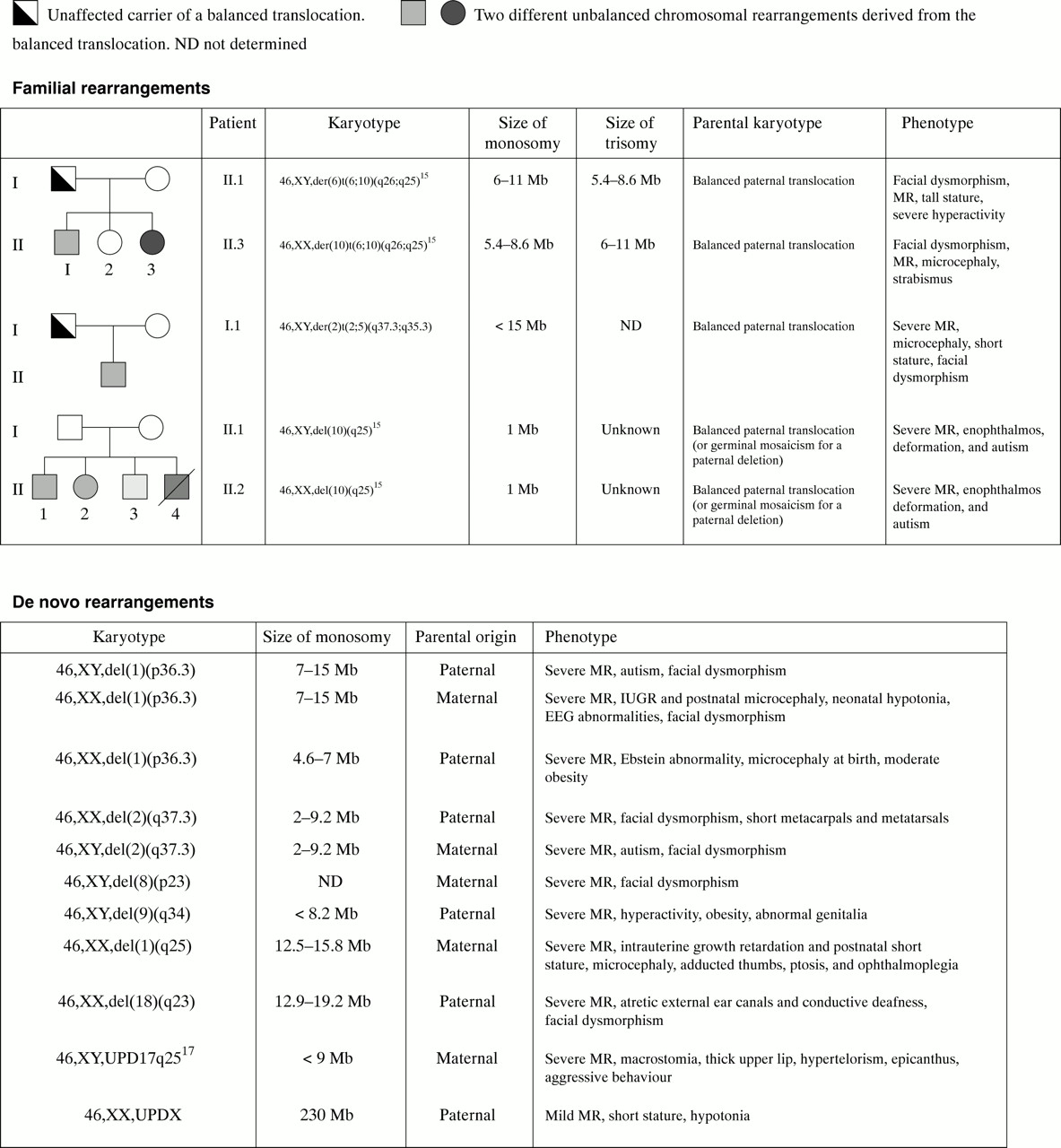
In order to confirm these findings and estimate the extent of the chromosome imbalance, we carried out genotyping analysis of the entire region and were able to confirm the following 15 anomalies: 1pter (three cases), 2qter (three cases), 6qter (two cases), 9q (one case), 10qter (two cases), 11qter (one case), 17qter (one case), 18qter (one case), and Xq (one case). Quantitative PCR allowed us to confirm two additional deletions of chromosome 10qter, while the remaining anomalies could not be confirmed by either genotype or FISH analyses. Altogether, we identified subtelomeric anomalies in 16/150 children (fig 3). Among them, 14 corresponded to deletions or duplications ranging in size from 1 to 19 Mb.
{kind=link}
{kind=link}
{kind=link}
Extent, parental origin, and associated phenotypes of chromosomal rearrangements.
In two cases (families 39 and 53), we confirmed an abnormal segregation of a marker with two alleles originating from the same parent and no contribution of the second parent. Further analysis showed that the affected child (family 53) carried a partial maternal heterodisomy of chromosome 17q25.3.17 In family 39, the child carried a paternal isodisomy of the entire X chromosome (table 1).
Genotype analysis with X chromosome markers. Markers used are shown on the left and sizes of the different alleles are given in base pairs for the parents and the affected child. Genotypes informative for monosomy/trisomy or maternal disomy are indicated in bold
We eventually studied the parents of all children carrying subtelomeric rearrangements by FISH analysis using probes of the rearranged region. In 9/14 cases, the rearrangement occurred de novo since parental chromosomes were normal. The rearrangement was of paternal origin in 5/14 cases and of maternal origin in 4/14 cases. In 5/14 families, the rearrangement was the result of the unbalanced segregation of a balanced paternal translocation. Careful retrospective analysis of patients' and parents' karyotypes using high resolution banding techniques confirmed the presence of the familial paternal translocation t(2;5)(q37.3;q35.3) and the de novo del(18)(q23) deletion, but failed to detect the other rearrangements.
Evidence for subtelomeric chromosome length polymorphism
In the course of this study, we observed five cases of telomere length polymorphism. In these cases, the child was missing one of the parental alleles at locus D17S1866, but not at the adjacent loci (D17S926, CEB49, and CEB84) (data not shown). In addition, the clinical features of these five cases were very different with respect to facial dysmorphism and growth anomalies (two cases of growth retardation and one case of advanced growth). It is therefore very likely that the terminal end of chromosome 17p is polymorphic in length and that deletion of the region corresponding to D17S1866 has no clinical relevance.
DISCUSSION
The present study shows that automated genotyping detected cryptic subtelomeric rearrangements in 10% of patients with idiopathic syndromic MR in our series. Since the first report by Flint et al,19 screening for cryptic subtelomeric rearrangements in idiopathic MR has yielded positive results in 5-7.4% of cases.10–12, 19–21 With a detection rate of 10.7%, this study supports the view that all children with severe idiopathic MR should be tested for telomere integrity. The efficiency, moderate cost (about $80 per family), and possible automatisation of the test make genotyping very effective for future large scale screening programmes.
The observation that UPD contributed to a significant proportion of idiopathic MR in our series is also important. Indeed, we ascertained two cases of UPD in our series. Since MR in families with two or more affected children is unlikely to result from UPD, the prevalence of this mechanism should average 1.7% (2/118 children tested). The phenotypic consequences of UPD are determined by mosaicism, genomic imprinting, non-mendelian inheritance of monogenic disorders, or by a combination of all these mechanisms and it is sometimes difficult to define the impact of each of these factors. Nevertheless, it appears that the systematic search for UPD in mentally retarded children may be a worthwhile approach to identifying hitherto unknown regions of genomic imprinting.
One should bear in mind, however, that genotyping has several limitations. First, this method requires access to DNA samples from both parents, which can be difficult to achieve in some cases. Second, because gene dosage cannot be reliably detected, small tandem duplication may have been overlooked in our survey. Third, the efficiency of this strategy is completely dependent on the informativeness and position of the markers used. The set of markers described here have an average heterozygosity score of 0.75 so that our strategy (with the analysis of a second marker in case of uninformativeness) can detect about 93% of monosomies and 68% of trisomies. Finally, we occasionally found abnormal segregation of single markers, but we were unable to confirm or exclude these findings in seven cases. This raises the question of whether we are dealing with false positive results owing to the instability of the microsatellites or very small deletions. While small deletions (130 kb) can cause MR,22 the relevance of such small anomalies will be difficult to determine unless additional cases are identified. Moreover, one must be cautious when interpreting abnormal segregation of markers since telomere length polymorphism can be involved, as we observed for the end of chromosome 17p.
The clinical presentation of several subtelomeric deletions is now well characterised, particularly monosomy 1pter,23 1qter,24 and 2qter.25 The present study suggests that terminal deletion of chromosomes 9q and 10q are most probably associated with hitherto undescribed MR syndromes. Because the patients presented with relatively specific features (obesity and behavioural problems for 9q34.3 deletion, and enophthalmos, foot deformation, and autism for deletion 10q26), careful analysis of the clinical features in patients carrying these rearrangements will hopefully help in recognising novel MR syndromes.
Based on the clinical presentation of subtelomeric anomalies, several groups have tried to define clinical criteria that would help to identify which patients should be studied. De Vries et al26 suggested that prenatal onset growth retardation and a familial history of MR are good indicators for subtelomeric defects.26 Our study suggests that congenital anomalies, behavioural problems, and postnatal growth retardation were the most frequent associated features in our series of MR children, while intrauterine growth retardation and a family history of MR were less frequent. The question of whether this observation is significant or is the result of ascertainment bias remains unanswered.
As far as the type of chromosome rearrangement is concerned, we observed a non-random distribution of telomeres involved in our series. Indeed, chromosomes known to share large regions of homology and/or length polymorphisms (such as 2q and 8p) were over-represented. It is also worth noting that all three inherited rearrangements were of paternal origin while de novo events occurred in both sexes. The rate of genetic recombination is known to increase in the telomeric regions in both sexes. This is particularly true in males.27 If recombination rate was playing a role in the pathogenesis of subtelomeric rearrangements, then the majority of telomere translocations would be of paternal origin. Our data partly support this view as all familial translocations but not all de novo rearrangements were of paternal origin in this study.
Telomere screening is a first step towards the goal of analysing the entire genome for chromosomal rearrangements in MR. Considering that subtelomeric rearrangements accounted for 10.7% of cases, it is tempting to speculate that extending our genotyping strategy to 400 markers evenly distributed along the chromosomes will detect interstitial rearrangements responsible for a significant (although unknown) proportion of MR cases.
Acknowledgments
We would like to thank the families involved in this study for providing samples. This work was supported by Centre National de la Recherche Scientifique and by Fondation de France.