Article Text

Abstract
Background: Paragangliomas are rare and highly heritable tumours of neuroectodermal origin that often develop in the head and neck region. Germline mutations in the mitochondrial complex II genes, SDHB, SDHC, and SDHD, cause hereditary paraganglioma (PGL).
Methods: We assessed the frequency of SDHB, SDHC, and SDHD gene mutations by PCR amplification and sequencing in a set of head and neck paraganglioma patients who were previously managed in two otolaryngology clinics in the USA.
Results: Fifty-five subjects were grouped into 10 families and 37 non-familial cases. Five of the non-familial cases had multiple tumours. Germline SDHD mutations were identified in five of 10 (50%) familial and two of 37 (∼5%) non-familial cases. R38X, P81L, H102L, Q109X, and L128fsX134 mutations were identified in the familial cases and P81L was identified in the non-familial cases. Both non-familial cases had multiple tumours. P81L and R38X mutations have previously been reported in other PGL families and P81L was suggested as a founder mutation. Allelic analyses of different chromosomes carrying these mutations did not show common disease haplotypes, strongly suggesting that R38X and P81L are potentially recurrent mutations. Germline SDHB mutations were identified in two of 10 (20%) familial and one of 33 (∼3%) non-familial cases. P131R and M71fsX80 were identified in the familial cases and Q59X was identified in the one non-familial case. The non-familial case had a solitary tumour. No mutations could be identified in the SDHC gene in the remaining four families and 20 sporadic cases.
Conclusions: Mutations in SDHD are the leading cause of head and neck paragangliomas in this clinic patient series. SDHD and SDHB mutations account for 70% of familial cases and ∼8% of non-familial cases. These results also suggest that the commonness of the SDHD P81L mutation in North America is the result of both a founder effect and recurrent mutations.
- succinate dehydrogenase
- carotid body tumours
- 11q23
- genomic imprinting
- CB, carotid body
- PGL, paraganglioma
- STRP, single tandem repeat polymorphism
Statistics from Altmetric.com
Head and neck, extra-adrenal and adrenal paraganglia comprise a diffuse neuroendocrine system with similar embryogenesis and histology and are dispersed from the middle ear and the skull base to the pelvic floor. Whereas the paraganglia in the head and neck region are located in close association with the parasympathetic nervous system along the cranial nerves and the arterial vasculature, the adrenal medulla and other extra-adrenal paraganglia are associated with the sympathetic nervous system (that is, the sympathoadrenal system).1 Paraganglioma refers to rare and mostly benign tumours that arise from any component of this neuroendocrine system. In the head and neck region, the carotid body (CB) is the largest of all paraganglia and is also the most common site of the tumours. Other tumour locations in the head and neck include jugulotympanic, vagal, laryngeal, aorticopulmonary, and minor locations such as the orbit and the thyroid.2
The aetiology of head and neck paragangliomas has been linked to the chemoreceptor function of the normal paraganglia.3 There is an increased incidence of the tumours in people living at high altitude, presumably caused by the reduced atmospheric oxygen levels. Certain medical conditions with chronic arterial hypoxaemia are also thought to be associated with hyperplastic growth of paraganglia.4 In the absence of chronic hypoxic stimuli, paragangliomas often develop as a result of a genetic predisposition. At least four genetic loci have been implicated in the pathogenesis of hereditary paraganglioma (PGL). Three of the loci, PGL1 on chromosome 11q23,5 PGL3 on chromosome 1q21,6 and PGL4 on chromosome 1p36,7 encode the mitochondrial complex II (succinate dehydrogenase, succinate:ubiquione oxidoreductase) subunits SDHD,8SDHC,9 and SDHB,7 respectively. The PGL2 gene at 11q1310 has yet to be confirmed and identified. PGL1 is transmitted as an autosomal dominant trait with age dependent penetrance liabilities when transmitted through fathers, whereas maternal transmission does not cause tumour development.11 This parent of origin effect, which is most consistent with genomic imprinting, suggests that expression of SDHD is altered through sex specific epigenetic modifications during gametogenesis, although the exact molecular mechanisms remain unknown. Interestingly, so far only maternal transmissions have been observed in the limited number of PGL families with SDHC6 and SDHB7 mutations.
Identifying whether paraganglioma is heritable in a patient is of utmost significance for clinical management. The proportion of heritable cases has been variably estimated from 10%12 to as high as 50%.11 A recent analysis of an unbiased sample of clinic patients with head and neck paragangliomas uncovered the presence of a positive family history in ∼25% of patients.13 When heritability was defined as the presence of a positive family history or tumour multifocality, this ratio increased to ∼35% in the same sample. The identification of the underlying genes enables us to explore the molecular basis of this heritability, as the relative role of each of the mitochondrial complex II subunits is unknown. In a recent study, SDHD mutations were identified in 30 of 32 (∼94%) PGL families and in 20 of 55 (∼36%) isolated cases in The Netherlands.14 Twenty-four of the 32 (75%) Dutch PGL families were the result of a single founder mutation. However, given the genetic (locus) and non-genetic heterogeneity in the aetiology of paragangliomas, it is unknown whether SDHD plays such a prominent role in other populations. It is conceivable that a fraction of paraganglioma patients without a family history could be carriers of germline SDHD mutations, because of imprinting and age dependent penetrance that obscure the familial nature of the PGL1 tumours. Accordingly, previous analyses of patients without a family history of head and neck paragangliomas14 and those with phaeochromocytomas (that is, adrenal paragangliomas)15 showed germline mutations in the SDHD gene. To determine the relative frequencies of SDHD, SDHC, and SDHB mutations in the aetiology of head and neck paragangliomas, we performed mutation analysis in a group of patients ascertained from two clinic patient populations in the United States.
MATERIALS AND METHODS
Head and neck paraganglioma patients
The subjects were patients diagnosed with head and neck paragangliomas, originating in the carotid body, jugular foramen, vagal nerve, temporal bone, or middle ear, at the Department of Otolaryngology at the University of Pittsburgh School of Medicine, Pittsburgh, PA and at the House Ear Institute, Los Angeles, CA. The recruitment of the subjects was performed either for the ongoing PGL linkage studies16, 17 or through a more recent study aimed at establishing the proportion of heritable cases of paragangliomas by clinical criteria.13 The latter study followed a procedure that involved mail questionnaires and structured telephone interviews and has been described in detail elsewhere.13 Family 33 has been independently ascertained from Canada and only one family member was available for the research study. Both studies were approved by the Institutional Review Board of the University of Pittsburgh.
Classification of subjects on the basis of family history
The subjects were classified as familial if two or more subjects of an extended pedigree were diagnosed with paragangliomas as documented either by hospital records or by the family history reported in the questionnaires. On the basis of positive family history, 18 subjects were classified into 10 families. A total of 37 subjects who were recruited to the study had no family history. Five of the 37 non-familial cases had multiple tumours. Pedigrees for families 1, 3, 4, 9, and 13, which had multiple affected subjects, have been described previously.16 Maternal transmission was observed in family 9. None of the five new families (fig 1) showed maternal disease transmission.

The new PGL families constructed from the subjects with head and neck paragangliomas. Affected subjects available for the study are shown with an asterisk. Maternal transmission is not observed in the pedigrees. The unaffected subjects' symbols in each pedigree have been collapsed to preserve anonymity. The subject depicted with a question mark in family 27 was reported to have a small solitary mass in the skull base by radiological analysis. However, this subject does not carry the SDHD mutation (table 1) detected in her affected relatives and is under further cliinical investigation.
Genotyping and haplotype analysis
DNA was isolated either from peripheral blood by standard phenol-chloroform extraction (PGL linkage study) or from cheek swabs using a commercial kit (Puregene D-500A) following the manufacturer's protocol (clinical study). The genotyping was performed only in the DNA obtained from peripheral blood. The simple tandem repeat polymorphisms (STRPs) used in genotyping and the haplotype analysis have been described elsewhere,18 except for the new polymorphic tetranucleotide repeat marker, D11S5030, which was located approximately 40 kb telomeric to the 3`-SDHD. This STRP had five alleles in 20 unrelated chromosomes with a calculated heterozygosity of 0.72. The primer sequences for D11S5030 that were used for the PCR amplification was 5`-CTA AAGGATTGAGTCATGCCCTT-3` and 5`-GACAAGAGTGAGAC CCTGTC-3` and allele sizes were 132, 134, 136, 140, and 144 bp. All PCR amplifications and genotypings were performed under standard conditions as described previously.17 The order and the distance information for the STRPs have been obtained by the analyses of fully sequenced regional genomic clones in Genbank. D11S5016, D11S5025, D11S5027, and D11S5028 map to Genbank Accession No AP001781 (181 kb), D11S5017 and D11S5015 map to Genbank Accession No AP000907 (88 kb), and D11S5019 and D11S5030 map to Genbank Accession No AP002007 (169 kb). The AP000907 sequence bridges the AP002007 and AP001781 sequences as detected by BLAST analysis of its sequence ends. The distance between D11S1347 and D11S5030 was estimated by electronic contig mapping of AP002007 onto a regional BAC clone (B162L1, 130 kb) and AB042297 (85 kb), both of which contain D11S1347.
Mutation analysis
SDHD mutation analysis was performed by SSCP analyses and direct sequencing as described previously.8 For the DNA extracted from peripheral blood, the exons were amplified directly using the previously described primers. Because of the limited amounts of DNA obtained from cheek swabs, the DNA was preamplified before gene analysis. For SDHD, the exons were first preamplified for 30 cycles using external primers under standard conditions. The external primers used in the first round amplifications for each exon were as follows: exon 1, F: 5`-TCGTCGTCGTGGGTGGGAA-3` and R: 5`-CTGGCT GGAGGCTACGCTA-3`; exon 2, F: 5`-CAGTCCTGTTAAAGG AGAGGT-3` and R: 5`-CCCTACAGGTAGGAAGTCCT-3`; exon 3, F: 5`-GATGTGTGTTTCTCACATCAA C-3` and R: 5`-CATTTC AATCAACTTCTCCCTCA-3`; exon 4, F: 5`-CAGCCAAGTTATC TGTATAGTCT-3` and 5`-GCAGAGGCAAAGAGGCATAC-3`. The preamplified samples were diluted 20-fold and subsequently amplified with the originally described primers.8
To sequence the six coding exons of SDHC and the eight coding exons of SDHB, the DNA extracted from the cheek swabs was first preamplified using Degenerate Oligonucleotide PCR (DOP) technique following a commercial protocol (Roche Molecular Biochemicals). The DOP PCR amplification was performed for 50 cycles. Each cycle was composed of the following steps: one minute at 94°C, two minutes at 37°C, four minutes at 55°C, 30 seconds at 68°C. The reaction was finalised with five minutes' incubation at 74°C. An aliquot of the DOP PCR product was subsequently used to amplify the gene exons. The primer sequences for SDHB exon 1 and exon 2, and the amplicon sizes were F: 5`- GGAGAGCGACCTCGGGGTT-3`, R: 5`-GTCTCTGTGGCTTTCCTGACT-3` (170 bp) and F: 5`-CCA GCAAAATGGAATTATCTTGT-3`, R: 5`-CTCTCCTTCAATAGCTG GCTT-3` (233 bp). SDHB exons 3-8 were amplified using previously reported primers.7
The primers used to sequence the SDHC exons and their amplicon sizes were as follows. 1F: 5`-CACTTCCGTCCA GACCGGAA-3`, 1R: 5`-ACCCAGACAGCGCCCACTCA-3` (516 bp) 2F: 5`-GTGTTTGATTAACTCTATTTTGCAT-3`, 2R: 5`-CTA TTGCTCTTCCCTAAGGAA-3` (645 bp); 3F: 5`-TTCTCCAT GTTGGTCAGGCT-3`, 3R: 5`- CTGGCTCCAGAATCCTTCCT-3` (327 bp); 4F: 5`-GTGCCTATTTCAGAATTAGTTT-3`, 4R: 5`-GAA TCTGAGCACAGTGCAAACT-3` (346 bp); 5F: 5`-GCTGTGAC AAGCTACTTGGT-3`, 5R: 5`-TGTGCAAATCCCGAATTAACT-3` (219 bp); 6F: 5`- GCGCTTTTCTCTAGAATCATG-3`, 6R: 5`-CCCAGGGCAGAAGCCACAGAGCT-3` (602 bp). The different number of non-familial cases analysed for each gene reflects the fact that the coding region of SDHB and SDHC could not be entirely sequenced in some non-familial cases owing to the poor quality of DNA preamplified from cheek swab DNA.
RESULTS
Mutation analyses of the paraganglioma patients
Mutations in SDHD were detected in five of the 10 (50%) PGL families and two of the 37 (∼5%) subjects without any family history (table 1). The identified mutations were predicted to truncate or dramatically alter the conformation of the SDHD protein product, cybS. Family 3 and the disease causing mutation H102L have been reported previously.8 The mutations in family SP64 and family 27 introduce premature stop codons that remove most of exon 4 from the mature cybS. R38X and P81L were identified in family 1 and family SP52, respectively. Subjects SP59 and SP77 who had no family history also carried the P81L mutation. Both subjects had multiple tumours in the head and neck region. The mutations R38X and P81L were previously reported in other families linked to the 11q23 locus8, 19 and their potential impact on cybS has been discussed.
Mutations identified in SDHD and SDHB
Mutations in SDHB were identified in two of the 10 (20%) PGL families and in one of the 33 (∼3%) subjects (including the two cases with SDHD P81L mutations) without any family history. P131R and M71fsX80 mutations were identified in families 9 and 13,16 respectively. Both mutations segregated with the disease phenotype in three affected subjects in family 9 and in two affected subjects in family 13. The P131R mutation alters an amino acid residue conserved among five eukaryotic species and was not detected in 200 normal chromosomes by PCR amplification of exon 4 and TaqI restriction enzyme digestion, which recognises the mutant allele. The M71fsX80 mutation of family 13 is predicted to cause a very early truncation of the SDHB protein product. A premature stop codon mutation, Q59X, was identified in one subject without a positive family history. This subject did not have multiple paraganglioma tumours.
No mutations could be identified in the SDHC gene in the remaining PGL families 4, SP48, SP56, and in 20 subjects (including the three cases with multiple tumours) who had no family history and no mutations in the SDHD and SDHB genes. Availability of multiple affected subjects in family 4 enabled us to show that the markers at chromosome 11q23 and chromosome 1p36 did not segregate in the affected subjects, thus confirming the exclusion of the SDHD and SDHB genes by linkage. However, several markers at the SDHC containing region on chromosome 1q21 showed cosegregation with the disease phenotype, thus precluding the exclusion of SDHC by linkage (data not shown). Additional members of families SP48 and SP56 were not available to assess the segregation of the three PGL loci further.
Haplotype analyses of the disease chromosomes carrying the P81L and R38X mutations
Previously, haplotype analyses had shown extensive haplotype sharing in four families with P81L mutations, suggesting the presence of a common ancestral mutation in these US families.8–17 The same mutation was subsequently identified in three other families from the USA,19 one family from the UK,14 and one family from Australia.20 Similarly, the R38X germline mutation was independently reported in two families from the USA8, 14 and in one subject with extra-adrenal paraganglioma.15 To test whether P81L and R38X cases were the result of founder mutations, we performed haplotype analyses using polymorphic markers located very close to the SDHD gene.
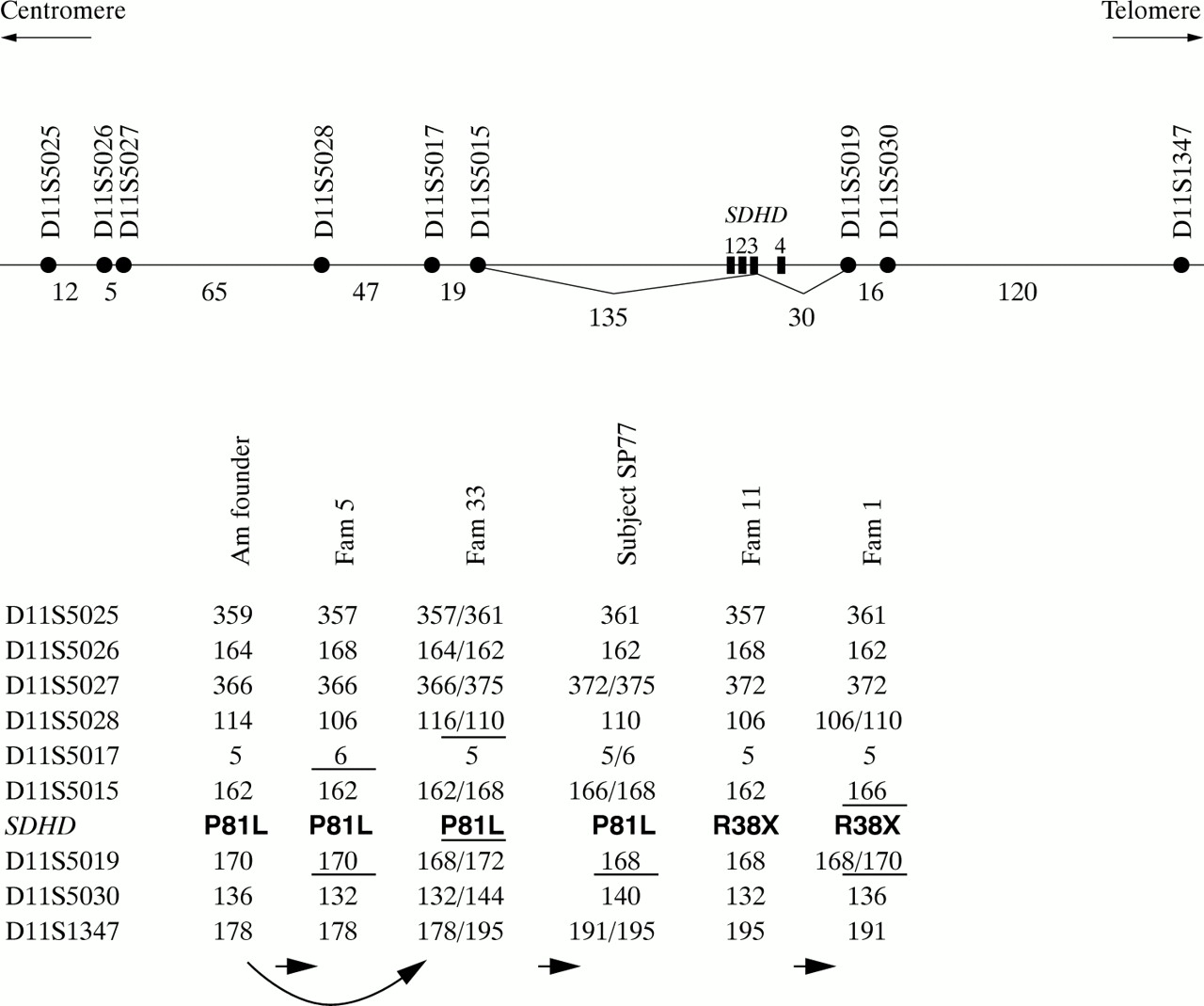
A visual inspection of the haplotypes comprising nine STRPs distributed within an approximate 450 kb region around the SDHD gene did not show a single haplotype for each mutation (fig 2). The identical alleles observed for the markers which immediately flank the SDHD gene were common among normal control chromosomes: D11S5015 allele 162 had a frequency of ∼35% and D11S5019 alleles 168 and 170 had frequencies of ∼50% and 45%, respectively. Therefore, these alleles shared among families cannot be readily concluded to be identical by descent. Even under the conservative assumption that the observed alleles are identical by descent, five independent ancestral recombination events within an approximate 200 kb interval between D11S5017 and D11S5030 would be required to explain the P81L haplotypes observed in the American founder families, family 5, family 33, and subject SP77. Similarly, the hypothesis that family 118 and family 1 derive from a common ancestral haplotype would require the occurrence of two recombination events. Previous analysis of a total of 632 meioses within an approximately 2 Mb interval between D11S897 and D11S1647, a region that also contains the SDHD region analysed here, uncovered 11 recombination events (0.87 cM/1 Mb) without any significant sex specific difference.21 Assuming random distribution of the recombination breakpoints within this interval, the probability of a recombination event occurring within the 200 kb interval tested in this study is estimated to be 0.87/5 = ∼0.17%. Thus, the very low probability of a single recombination event within this small interval strongly suggests that the observed haplotypes do not originate from single ancestral haplotypes, although the possibility of very old mutations cannot be entirely excluded.

{kind=link}
{kind=link}
Haplotype analyses of the chromosomes with P81L and R38X mutations. The physical map of the markers and approximate distances between them are shown in kilobases at the top. The American founder haplotype was previously observed in four unrelated families.8, 17 For the P81L families, the arrows indicate a pathway in which the observed haplotypes derive from the American founder haplotype by recombination events occurring in the intervals indicated by the horizontal bars. This scenario would require the occurrence of five recombinations. Similarly, if family 11 and family 1 were identical by descent for the R38X mutation, two recombinations must have occurred.
DISCUSSION
To determine the prevalence of SDHB, SDHC, and SDHD germline mutations in the aetiology of head and neck paragangliomas, we tested a total of 18 subjects (10 families) with a family history and 37 subjects without a family history, all of whom were previously evaluated in two otolaryngology clinics. We identified mutations in SDHD in five of the 10 (50%) families and two of the 37 (∼5%) subjects without a family history. These two subjects were among the five cases that showed multifocality without a family history, suggesting that tumour multifocality is predictive of hereditary PGL. Mutations in SDHB were identified in two of the 10 (20%) PGL families and in one of the 33 (∼3%) subjects without any family history. The one non-familial case had a single tumour. No mutations were identified in the SDHC gene in the remaining four PGL families and in 20 non-familial cases without a family history and without mutations in the SDHD and SDHB genes. Thus, mutations in SDHD are the leading causes of paraganglioma tumours among the three mitochondrial complex II genes tested in this study. These findings also indicate that mutations in SDHD and SDHB account for 70% of head and neck paragangliomas with a positive family history and for ∼8% of those without a positive family history. The remaining familial cases could be the result of mutations that are not detectable by exon amplification and sequencing in the tested genes or there may be other genes involved in the aetiology.
Germline SDHD mutations were predicted to occur in non-familial paragangliomas because the familial nature of the tumours could be obscured by reduced penetrance and imprinting. In fact, the frequent (∼36%) detection of SDHD founder mutations among non-familial cases in The Netherlands14 indicates that some of the apparently sporadic cases do inherit their disease genes from their fathers. In our study, however, it was unclear whether P81L mutations in the two non-familial paraganglioma cases were inherited or arose de novo through the recurrent mutational mechanism. Other family members were not available to distinguish between these two competing hypotheses. We detected the P81L mutation in three new cases and the R38X mutation in one new case. Because both mutations had previously been reported and P81L was further implicated as a founder mutation among US families,17 we tested for the presence of a single founder chromosome for each mutation by the analyses of nine STRPs located within an approximately 450 kb region around the SDHD gene. Haplotype analyses showed no evidence for an extensive haplotype sharing for each mutation, strongly suggesting that P81L and R38X are potentially recurrent mutations in the SDHD gene. Both mutations result from CpG to TpG transitions that are likely to be triggered by the deamination of methylcytosine. We conclude that both a founder effect and recurrent mutations are likely to be responsible for the high prevalence of P81L in North America. Thus, the P81L mutation may be recommended as the first mutation to test by PCR and restriction enzyme analysis in patients with paraganglioma tumours.
The mutations in SDHB and the pedigrees in which they are identified contrast with those of SDHD. Six different mutations have now been described in SDHB without evidence for a founder mutation. This finding contrasts with SDHD gene mutations that show strong founder effects. Taschner et al14 found that two founder mutations in the SDHD gene accounted for 30 of 32 PGL families in The Netherlands and we found that the P81L founder mutation accounts for more than half of the PGL1 linked families ascertained in the USA.22 Accordingly, the pedigrees with the SDHB mutations are relatively small with only a few affected subjects. This finding also contrasts with the presence of many extended SDHD mutant families which had enabled linkage mapping of the PGL1 locus before the gene was identified.5, 26 Thus, under the assumption that the de novo mutation rates of the two genes are comparable, the mutations in SDHD may be associated with better phenotypic fitness than those in SDHB. The presumed fitness difference between the two genes can be explained in part by the genomic imprinting at SDHD which effectively reduces the overall disease penetrance. Unlike SDHD, the presence of paternal transmission in family 13 and maternal transmission in family 9 suggests that the inheritance of SDHB does not show any parent of origin effects.
Finally, the contribution of SDHC to the aetiology of paragangliomas remains unconfirmed. No coding mutations were discovered in SDHC. However, the chromosome 1q21 genomic region where SDHC resides could not be excluded by linkage in one of the multiplex families. This finding suggests that either unconventional mutational mechanisms, which could not be detected by the methods we used in this study, are operative in SDHC or that there is a distinct nearby gene that also contributes to the aetiology of PGL. Further studies are needed to suggest additional genes that also contribute to the genetics of hereditary paragangliomas.
Acknowledgments
We thank all the patients who participated in this study. This work was partly supported by grant 5 P60 DE13059 from the National Institute of Dental and Craniofacial Research to the Oral Cancer Center at the University of Pittsburgh.