Article Text

Abstract
OBJECTIVE To review all cases with segmental and/or complex uniparental disomy (UPD), to study aetiology and mechanisms of formation, and to draw conclusions.
DESIGN Searching published reports in Medline.
RESULTS The survey found at least nine cases with segmental UPD and a normal karyotype, 22 cases with UPD of a whole chromosome and a simple or a non-homologous Robertsonian translocation, eight cases with UPD and two isochromosomes, one of the short arm and one of the long arm of a non-acrocentric chromosome, 39 cases with UPD and an isochromosome of the long arm of two homologous acrocentric chromosomes, one case of UPD and an isochromosome 8 associated with a homozygous del(8)(p23.3pter), and 21 cases with UPD of a whole or parts of a chromosome associated with a complex karyotype. Segmental UPD is formed by somatic recombination (isodisomy) or by trisomy rescue. In the latter mechanism, a meiosis I error is associated with meiotic recombination and an additional somatic exchange between two non-uniparental chromatids. Subsequently, the chromatid that originated from the disomic gamete is lost (iso- and heterodisomy). In cases of UPD associated with one isochromosome of the short arm and one isochromosome of the long arm of a non-acrocentric chromosome and in cases of UPD associated with a true isochromosome of an acrocentric chromosome, mitotic complementation is assumed. This term describes the formation by misdivision at the centromere during an early mitosis of a monosomic zygote. In cases of UPD associated with an additional marker chromosome, either mitotic formation of the marker chromosome in a trisomic zygote or fertilisation of a gamete with a marker chromosome formed in meiosis by a disomic gamete or by a normal gamete and subsequent duplication are possible.
CONCLUSIONS Research in the field of segmental and/or complex UPD may help to explain undiagnosed non-Mendelian disorders, to recognise hotspots for meiotic and mitotic recombinations, and to show that chromosomal segregation is more complex than previously thought. It may also be helpful to map autosomal recessively inherited genes, genes/regions of genomic imprinting, and dysmorphic phenotypes. Last but not least it would improve genetic counselling.
- genomic imprinting
- isochromosome
- Robertsonian translocation
- uniparental disomy (UPD)
Statistics from Altmetric.com
Uniparental disomy (UPD) describes the inheritance of both homologues of a pair of chromosomes from only one parent.1Either the presence of both homologues (“heterodisomy”), or of two copies of one homologue (“isodisomy”), or a mixture of both are possible, reflecting the number and localisation of meiotic recombinations. Mechanisms of formation are trisomy rescue, gamete complementation, mitotic duplication, and postfertilisation error.2 Problems associated with UPD are (1) placental or even fetal mosaicism mostly because of formation by trisomy rescue, (2) homozygosity of autosomal recessively inherited mutations, and (3) aberrant genomic imprinting describing parent of origin dependent gene expression. Well known examples are maternal UPD(15) in approximately 25-30% of patients with Prader-Willi syndrome (PWS, MIM 176270) and paternal UPD(15) in approximately 2-3% of cases with Angelman syndrome (AS, MIM 105830).3 Apart from PWS and AS, more than 100 cases of UPD of other chromosomes have been reviewed recently.4 All these cases describe UPD of a whole chromosome. In contrast, apart from paternal UPD(11)(p15→pter) in 10-20% of cases with Beckwith-Wiedemann syndrome (BWS, MIM 130650),5 UPD of segments of chromosomes or UPD associated with a complex chromosomal rearrangement have rarely been reported.
Previous published reports of segmental and complex UPD using molecular investigations are reviewed here. The aetiology and mechanisms of formation are discussed and some conclusions are drawn. Segmental UPD is defined as UPD of a part of one chromosome (interstitial or telomeric) together with biparental inheritance of the rest of this pair of chromosomes and a normal karyotype (fig 1A, B). Complex UPD is subdivided into three groups: (1) UPD of a whole chromosome associated with a “simple” translocation (fig 1C) or a Robertsonian translocation of two non-homologous acrocentric chromosomes (fig 1D); (2) UPD associated with isochromosomes of the short arm and the long arm of a non-acrocentric chromosome (fig 1E, F) or with a Robertsonian translocation between homologous acrocentric chromosomes (fig 1G); and (3) “sensu strictu” complex UPD defined as UPD of a part or of a whole chromosome directly involved in or associated with a structural and/or numerical chromosomal complement other than (A) and (B) (fig 1H).

Diagrams of segmental and complex UPD: (A) telomeric segmental UPD, (B) interstitial segmental UPD, (C) UPD associated with a “simple” translocation, (D) UPD associated with a translocation of non-homologous acrocentric chromosomes, (E) pat/mat UPD associated with two isochromosomes, one of the short arm and one of the long arm of a non-acrocentric chromosome, (F) UPD associated with two isochromosomes of the same parental non-acrocentric chromosome, (G) UPD associated with a translocation between two homologous acrocentric chromosomes, and (H) “sensu strictu” complex UPD in the instance of an additional marker chromosome.
Review
SEGMENTAL UPD ASSOCIATED WITH A NORMAL KARYOTYPE (TABLE1)
Mosaicism for paternal UPD of the chromosomal segment 11p15→pter is found in approximately 10-20% of cases with BWS.5 A complex interaction between two imprinting centres regulating up- and downexpression of the paternally expressedIGF2 gene as well as the maternally expressed H19,p57 KIP2, andKVLQT1 genes, all located within 11p15.5 (and other still unknown genes), is considered to be aetiologically relevant.6
Segmental uniparental disomy associated with a cytogenetically normal karyotype
Pure segmental UPD of other chromosomes not associated with a cytogenetically abnormal karyotype is extremely rare. To the best of my knowledge, only nine cases have been reported so far. The first is a 22 year old healthy woman with interstitial maternal isodisomy 2p16 shown by two markers homozygous in the child and heterozygous in the mother.7 The second was detected by loss of heterozygosity in one case in a study of linkage of DIDMOAD syndrome (MIM 222300) to the short arm of chromosome 4.8 No symptoms other than those typically found in DIDMOAD syndrome were present. Breakpoints were not reported precisely. Two other cases with segmental UPD were found by chance. The first with maternal isodisomy 4q21→q35 was ascertained in a patient with abetalipoproteinaemia resulting from a homozygous intron 9 splice acceptor G(−1) to A mutation.9 A minimal region of UPD was shown by eight informative short tandem repeat markers spanning a region of approximately 150 cM. The second case showed paternal uniparental isodisomy 6p ascertained by homozygosity for a mutation in the steroid 21-hydroxylase gene and reduction to homozygosity in eight microsatellite markers located on the short arm of chromosome 6.10 Three markers located on 6q were inherited biparentally. Paternal uniparental isodisomy 6q24→qter was found in a male newborn with neonatal diabetes, decreased subcutaneous tissue, and a patent ductus arteriosus. Craniofacial dysmorphism included a prominent occiput, lambdoidal ridging, a small fontanelle, shallow orbits, a prominent nose, dysmorphic ears, gingival and labial hypertrophy, macroglossia, a high palate, and micrognathia.11 In screening for maternal UPD(7) among patients with Silver-Russell syndrome (SRS), a 16 month old girl with maternal isodisomy 7q31→qter was found.12 Of specific interest are two cases with maternal segmental heterodisomy 14. The first was a 3½ year old girl with developmental delay, hypotonia/joint laxity, macrocephaly, and maternal heterodisomy 14q23→q24.2.13 The second was a patient with pre- and postnatal growth retardation associated with maternal heterodisomy 14q12→q24.3.14 Maternal UPD(Xq27→qter) was also described in a normal woman.15
UPD OF A WHOLE CHROMOSOME ASSOCIATED WITH A “SIMPLE” TRANSLOCATION OR A ROBERTSONIAN TRANSLOCATION (TABLE2)
Maternal uniparental heterodisomy of chromosome 7 was found in a case with a phenotype resembling SRS and a balanced 7;16 translocation (46,XX,t(7;16)(q11.2q22)).16 PWS resulting from maternal UPD(15) was described in a boy with an unbalanced maternal 3;15 translocation (47,XY,+der(15)t(3;15) (p25q11.2)).17 AS resulting from paternal UPD(15) was found in a girl with an unbalanced paternal 6;15 translocation (45,XX, t(6;15)(p25.3q11.1)),18 and in a boy with an unbalanced paternal 8;15 translocation (45,XY,−8,−15, +der(8)t(8;15)(p23.3q11)).19 In all three cases the deletion was small and presumably without clinical relevance.
Uniparental disomy of a whole chromosome associated with a “simple” translocation (non-homologous Robertsonian translocations included)
Maternal heterodisomy 16 was described in a male newborn with a balanced 10;16 translocation (46,XY,t(10;16)(q11.2q11.1)) and confined placental trisomy 16 mosaicism.20 The phenotype was characterised by intrauterine growth retardation (IUGR), a small ventricular septal defect, and minor dysmorphism.
The specific situation of a non-homologous Robertsonian translocation associated with UPD of one of the chromosomes involved was reported 17 times. Three cases with UPD studied by molecular methods and associated with a non-homologous Robertsonian translocation were not considered, because no information on parental origin was given.21 22The majority of cases comprised maternal UPD, particularly maternal UPD(14) associated with a 13;14 translocation, five times de novo23-27 and four times familial.28-31Moreover, familial 13;14 translocations were found in one case with maternal UPD(13)32 and in two cases with paternal UPD(14).33 34 Only one case of maternal UPD(14) associated with a maternal 14;21 translocation is known.35PWS owing to maternal UPD(15) was reported in two families with maternal 14;15 translocations,36 37 in one family with a maternal 13;15 translocation,38 and, in addition, in one case with a de novo 13;15 translocation.39 To the best of my knowledge, there is no case of AS resulting from paternal UPD(15) associated with a Robertsonian translocation.
UPD OF A WHOLE CHROMOSOME ASSOCIATED WITH A CYTOGENETICALLY DEFINED ISOCHROMOSOME (TABLE 3)
UPD associated with an isochromosome and a correct amount of genetic material is possible (1) in the case of an isochromosome replacing the two homologues of a pair of acrocentric chromosomes (fig1G) or (2) by isochromosomes both of the short arm and of the long arm of a non-acrocentric chromosome (fig 1F). The latter has been described in five cases with isodisomic isochromosomes of both the short arm and the long arm of the same non-acrocentric chromosomes 1, 2, 4, and 9.40-44 Multiple abortions because of inadequate segregation were observed in all adults. The first was a 43 year old woman with short stature, ptosis, micro/retrognathia, myopathy, deafness, and a karyotype of 46,XX,i(1p)i(1q) associated with paternal isodisomy 1.42 The second was two cases with maternal isodisomy 2 and karyotypes of 46,XX,i(2p)i(2q). One of them was completely normal,40 while the other was affected by pre- and postnatal growth retardation, bronchopulmonary dysplasia, and minor anomalies.44 The cases with maternal UPD(4) and maternal UPD(9) were completely normal and ascertained by investigations initiated because of multiple abortions.41-43
Uniparental disomy associated with an isochromosome defined cytogenetically
In addition, two isochromosomes were reported in two cases with a maternal isodisomic isochromosome 7q and a paternal isodisomic isochromosome 7p, postnatal growth retardation, and dysmorphic features resembling SRS (fig 1E).45 46 The same karyotype was reported in a healthy woman with a maternal isodisomic isochromosome 2q and a paternal isodisomic isochromosome 2p.47
UPD of all acrocentric chromosomes has been described in variable numbers. One case with an isochromosome 14q and another case with an isochromosome 15q and UPD shown by molecular methods were not considered, because no information on parental origin was given.21 UPD of chromosome 13 associated with an isochromosome or a der(13;13) has been reported in seven cases. Twice there was maternal UPD(13),48 49 one of them in two generations,48 50 and five times there was paternal UPD(13).32 50-53 All patients are healthy. Of specific interest are isochromosomes 14q and 15q. Imprinted genes are located or assumed on both chromosomes. Six cases with PWS54-58 and six cases with AS,59-63 all with de novo isochromosomes 15q, have been recorded. Maternal UPD(14) has been described in eight cases27 35 57 64-68 and paternal UPD(14) in four cases,27 32 69 all with de novo isochromosomes 14q. The phenotype of maternal UPD(14) is characterised by pre- and postnatal growth retardation, early onset of puberty, advanced bone age, and minor dysmorphisms.70 In paternal UPD(14), polyhydramnios, skeletal anomalies, contractures, and dysmorphic features are typical findings.33 No problems or anomalies other than infertility were reported in two cases of maternal UPD(21),71 72 two cases of paternal UPD(21),57 73 one case of paternal UPD(22),74 and three cases of maternal UPD(22).75-77 All were associated with an isochromosome and apart from two of the latter75 76 were de novo.
A specific form of isochromosome was described by Piantanidaet al.78 The karyotype was 45,XX,−8,−8,+psu dic(8;8)(p23.3p23.3). Apart from maternal heterodisomy (8)(p23.3qter), there was a homozygous del(8)(p23.3pter). The patient was affected by ataxic gait, mental retardation, and dysmorphic features including microcephaly, a narrow forehead, bushy eyebrows and long eyelids, inner epicanthic folds, convergent squint, and dysplastic ears.
UPD OF PART OR A WHOLE CHROMOSOME DIRECTLY INVOLVED IN OR ASSOCIATED WITH A COMPLEX STRUCTURAL AND/OR NUMERICAL CHROMOSOMAL COMPLEMENT (TABLE 4)
UPD of part or a whole chromosome and an associated structural and/or numerical chromosomal aberration of this chromosome other than simple trisomy was reported in a minimum of 21 cases. All were de novo.
Uniparental disomy of parts or of a whole chromosome associated with a complex chromosomal rearrangement (isochromosomes excluded)
The first was a girl with IUGR, transient neonatal diabetes mellitus, paternal isodisomy 6, and an additional maternal r(6) in 37 out of 50 cells investigated.79
Molecular investigations showed maternal uniparental isodisomy 7 in a girl with a pheotype resembling SRS and a 47,XX,UPD(7)mat, +r(7)pat/46,XX,UPD(7) mat karyotype.80
A pseudodicentric chromosome 8 with maternal heterodisomy (8)(p23.1qter), del(8)(p23.3pter), and del(8)(p23.1pter) was found prenatally.81 Fetal necropsy showed hypoplasia of the cerebellar vermis and dilatation of tubules of the left kidney.
Well known for a long time is the association of AS or PWS with a mosaic idic(15). However, UPD(15) has been proven by molecular investigations in only seven cases with PWS,37 82-86 and two cases with AS,86 87 all with an additional mosaic idic(15). One case with a mosaic idic(15) and UPD(15) shown by molecular methods was not considered, because no information on parental origin was given.22 UPD(15) can be assumed in several other cases of PWS or AS associated with an idic(15). However, these cases were reported before the relevance of the parental origin of chromosome 15 was known and del(15q12) is sometimes difficult to detect by cytogenetic investigations only.
Maternal UPD(16) has been reported in a boy with various dysmorphic features, developmental delay, and partial trisomy 16p mosaicism only.88 In this case, formation by trisomy first, translocation second, and uniparental disomy and partial trisomy third was inferred.
Paternal uniparental isodisomy of 20p13→qter was found in a child with a 45,XY,−20,−20,+ter rea(20;20)(p13p13) karyotype in lymphocytes.89 The child was affected by multiple abnormalities including an absent left ear with a small right ear remnant, microcephaly, congenital heart disease, and Hirschsprung disease. Karyotyping of fibroblasts showed a second cell line with trisomy 20. IUGR, developmental delay, and hyperactivity were described in a boy with maternal heterodisomy 20 and an additional mar(20) in 20 out of 50 cells investigated.90
An adjacent 1 segregation associated with maternal heterodisomy (22)(q11.2→qter) was observed in a girl with multiple anomalies, developmental delay, and a der(22) t(11;22)dn.91
Several cases of complex UPD(X) have been reported. Unique is a case of paternal isodisomy in a 13 year old boy with a 45,X karyotype and normal psychomotor development.92 His Turner-like phenotype included growth retardation, low set and deformed ears, short and wide neck, broad chest, widely spaced nipples, short metacarpal bones, and slight cubitus valgus. Molecular investigations showed a translocation of the terminal part of Yp to the short arm of the paternally inherited X chromosome (Xp22.3;Yp11). Three maternal and one paternal functional isodisomy of parts of the X chromosome have been described in four patients with Turner syndrome resulting from structurally abnormal and mosaic karyotypes and various degree of developmental delay.93-95 Differences in the ratio of mosaicism in various organs and/or in the length of the functionally active X chromosome were discussed as being responsible for the developmental delay. Moreover, there are several cases where UPD is likely, but not definitively proven.96
Discussion
This survey on complex and segmental UPD indicated a broad spectrum of both cytogenetically recognisable abnormal chromosomal complements and completely normal karyotypes. All are the result of chromosomal rearrangements resulting from either meiotic, mitotic, or meiotic and subsequent mitotic abnormal recombinations.
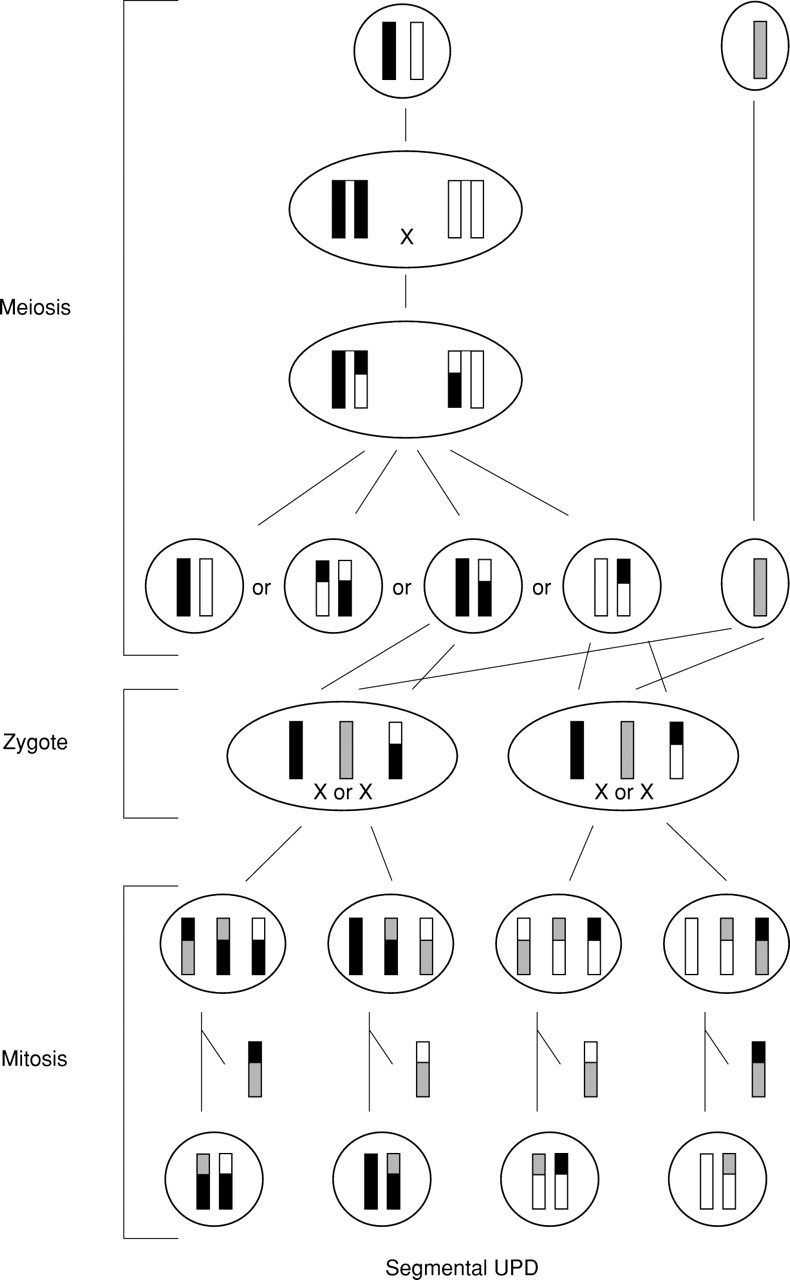
Segmental isodisomy other than paternal UPD(11)(p15pter) in Beckwith-Wiedemann syndrome was assumed to be formed postzygotically by a mitotic exchange between non-sister chromatids (fig2).9 11 However, then UPD should be present in each daughter cell, maternal UPD in one and paternal UPD in the other one. With molecular studies it is not possible to recognise such an exchange. The only exception would be the loss of the opposite UPD either because of occurrence in a very early mitosis and subsequent splitting in embryonic and extraembryonic tissues or, as assumed in Beckwith-Wiedemann syndrome, because of lethality of cells with maternal isodisomy. The latter could be excluded in paternal UPD(6p)10 and paternal UPD(6q24→qter)11 by two cases with complete maternal UPD(6).97 The same is true for maternal UPD(7q31→qter)13 by two cases with paternal UPD(7).98 Moreover, telomeric isodisomy might also be formed by trisomy rescue (fig 3). There, a meiosis I error is followed by a meiotic recombination and associated with a mitotic crossing over between one paternal and one maternal chromatid. Subsequently, the chromatid involved in the exchange and originating from the disomic gamete is lost. This scenario would also explain the molecular findings. Therefore, in segmental uniparental isodisomy not only a completely mitotic formation but also a meiotic/mitotic mechanism is possible. Isodisomy or heterodisomy depends on the occurrence of a meiotic recombination only. This is particularly important for evaluation of the clinical phenotype. In practice, the size of the isodisomic segment and the number of investigated markers heterodisomic in the parent and isodisomic in the patient should be considered as a clue towards meiotic/mitotic or purely mitotic formation. Trisomy rescue was assumed in a case with interstitial maternal heterodisomy 14q23→24.2.13 This case and a second one14 could be very helpful in localising one or more region(s) of genomic imprinting on chromosome 14. The same is true in the case with maternal UPD(7q31→qter).12 The phenotype, which was typical for SRS resulting from maternal UPD(7), narrowed the region of interest for mapping one or more SRS genes to approximately 40 Mb. However, a normal phenotype in people with segmental UPD despite an abnormal phenotype in the case of UPD of the whole chromosome would exclude this chromosomal region from genomic imprinting.
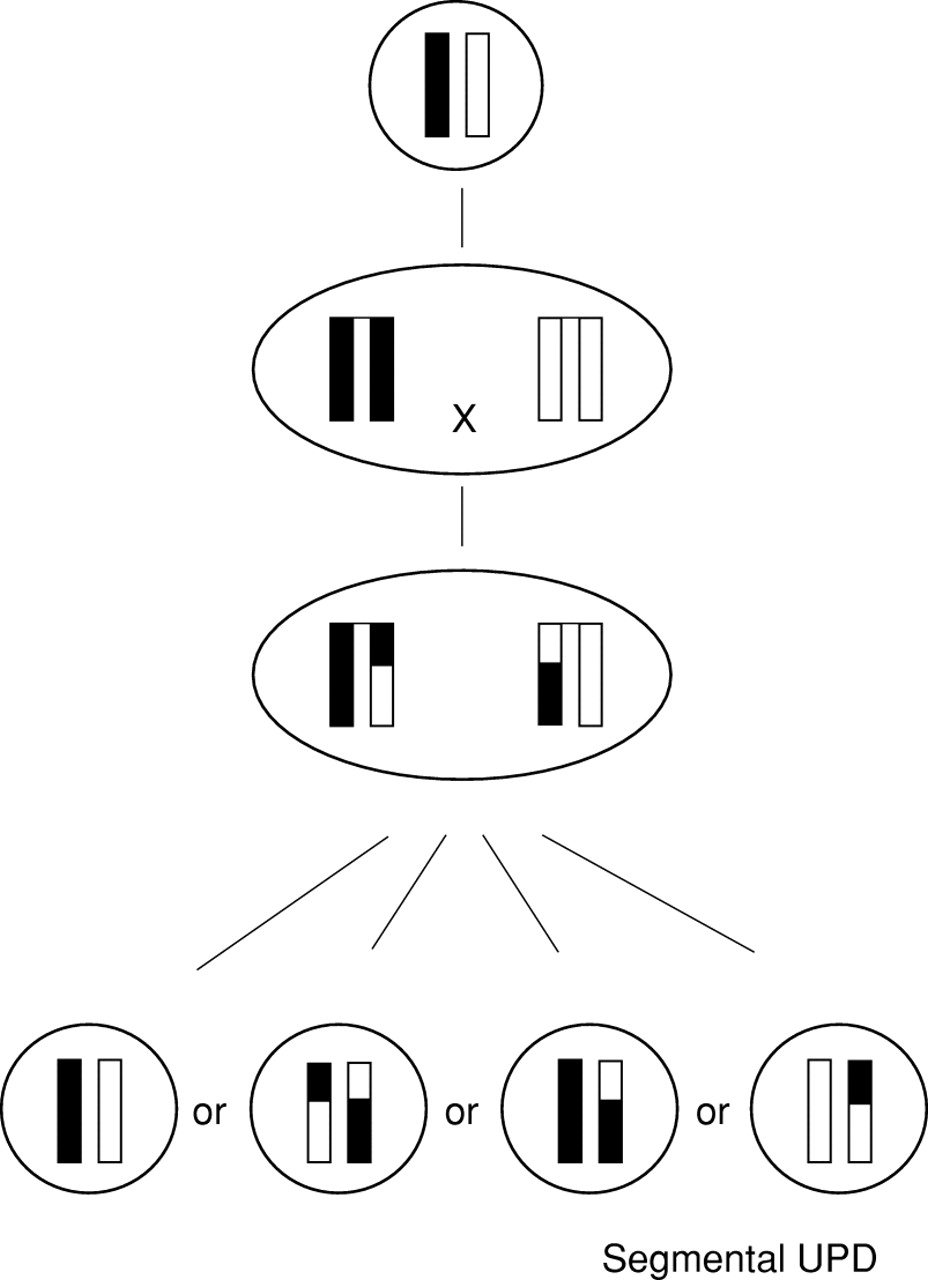
Mitotic formation of segmental UPD associated with a normal karyotype by somatic recombination between two non-sister chromatids and subsequent loss of both the inverse uniparental disomic and the biparental products.
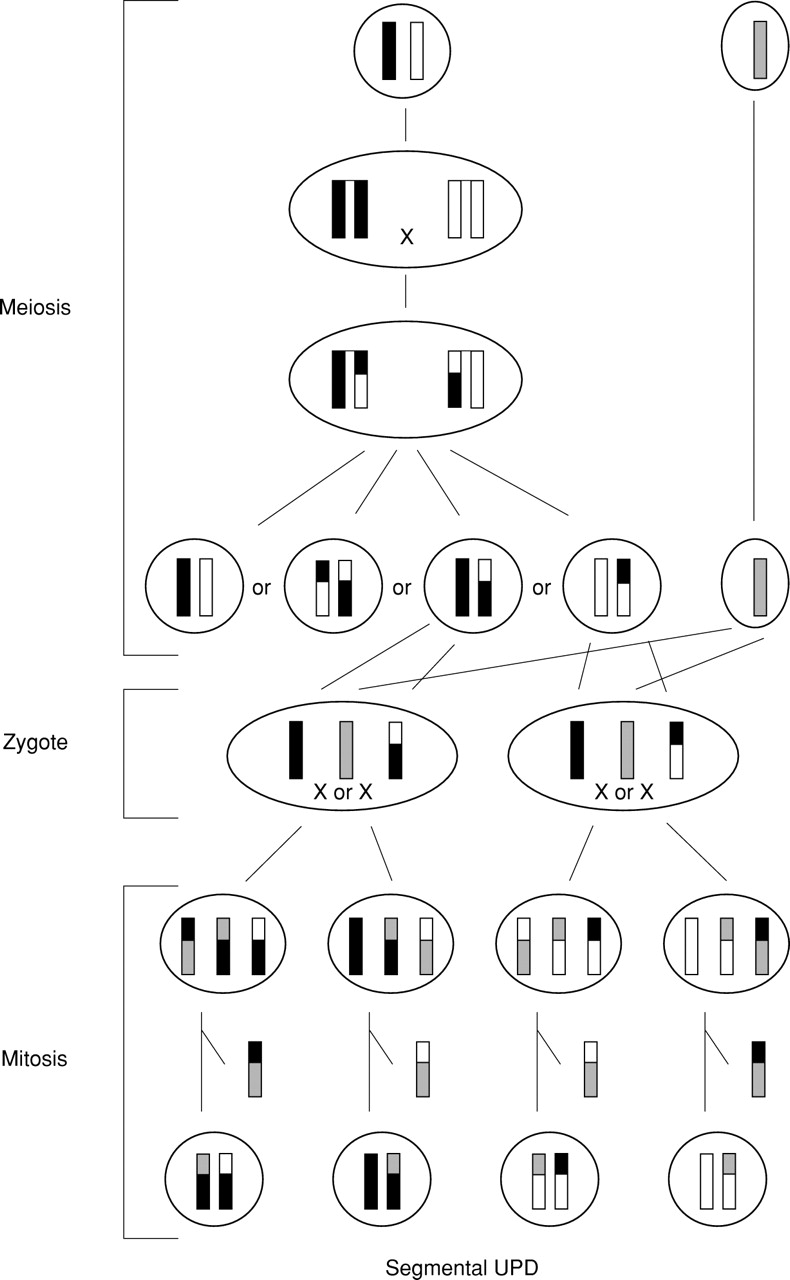
Formation of segmental UPD associated with a normal karyotype by trisomy rescue. A meiosis I error is associated with a meiotic recombination and followed by a mitotic crossing over between two non-uniparental chromatids. Subsequently, the chromatid involved in the exchange and originating from the disomic gamete is lost.
A major problem in ascertainment of cases with segmental UPD is the large number of molecular investigations required. Almost all cases reported so far were detected by chance. Investigations for UPD were only performed in cases with relevant phenotypic features and included only a few markers. Biparental inheritance of one or two markers is thought to be enough to exclude UPD. Screening for segmental UPD would require a panel of informative markers for chromosomal intervals sized not more than 10 cM. Nevertheless, in cases with a causally undefined phenotype such an approach would be worthwhile for several reasons. First, segmental isodisomy might be responsible for a relevant number of atypical cases of monogenic disorders or sporadic neoplasms. Recurrence risk would be low in the case of an autosomal recessively inherited disorder resulting from segmental UPD. Second, hotspots for meiotic and mitotic recombinations would be recognised. Third, cases with segmental UPD would be a powerful tool to map and to exclude autosomal recessively inherited genes, regions and genes of genomic imprinting, and dysmorphic phenotypes. The latter is of particularly interest, even in the era of sequencing the whole human genome, since a phenotype is more than a simple DNA sequence. Generally, segmental UPD would be of major interest both for clinical genetics and for uncovering the aetiology of many non-Mendelian disorders.
As shown in table 3, UPD has been described in eight cases associated with two isochromosomes, one of the long arm and one of the short arm of a non-acrocentric chromosome. All are isodisomic. In the five cases where both isochromosomes have the same parental UPD, this uniformity supports a mechanism of formation by misdivision at the centromere during the first or second mitosis of a monosomic zygote. Mitotic complementation would be an adequate term. In the case with paternal isodisomy 1 and a 46,XX,i(1p)(1q) karyotype, an aetiology other than genomic imprinting or confined placental mosaicism (CPM) is likely.42 The pattern of clinical findings is not typical for CPM. Furthermore, in one other case with paternal UPD(1) a normal phenotype was described.99 It may be that a (unknown) monogenic disorder resulting from homozygosity of an autosomal recessively inherited mutation is causative. This is less convincing in the two cases with maternal UPD(2) and a 46,XX,i(2p)i(2q) karyotype.40 44 Here, the clinical phenotype of pre- and postnatal growth retardation, bronchopulmonary dysplasia, and minor anomalies in the second case44 strongly resembles the anomalies described in cases with CPM.100
In the three cases where one isochromosome had maternal UPD and the other paternal UPD, a postzygotic formation is most likely.14 45 46 A misdivision at the centromeres of both homologous chromosomes in an early or even in the first mitosis of a normal zygote followed by the loss of each opposite short and long arm because of the lack of functioning centromeric material was assumed. Alternatively, but very unlikely, one isochromosome was already present in each gamete. The contrast between severe pre- and postnatal growth retardation in maternal UPD(7)101 and the only moderate postnatal growth retardation in maternal UPD(7q) and paternal UPD(7p)45 46 hint towards mapping of one or more imprinted genes.
In addition, there are variable numbers of chromosomes composed from two copies of the same acrocentric chromosome (table 3). In this context, one has to keep in mind that “isochromosome” is a cytogenetically defined term and describes a chromosome with two cytogenetically identical arms formed by a misdivision at the centromere or a U type exchange during meiosis.102 From a molecular point of view, a fusion of a maternal and a paternal homologue or of the two homologues from the same parent but with different alleles are Robertsonian translocations or derivative chromosomes. These differences are important to understand the clinical phenotype in the case of imprinted genes or homozygosity for autosomal recessively inherited mutations present in only one parent. The latter is not possible in heterodisomy, while genomic imprinting should work in isodisomy and heterodisomy equally. In contrast, both are unlikely in cases that were formed postzygotically. In theory, several different mechanisms of formation of UPD associated with an isochromosome are possible: (1) meiotic misdivision at the centromeres resulting in a gamete with an isochromosome, fertilisation by a normal gamete, and subsequent mitotic loss of the homologous chromosome (fig 4A); (B) meiotic misdivision at the centromeres and fertilisation by a nullisomic gamete (fig 4B); (C) mitotic misdivision at the centromeres of sister or non-sister chromatids in a trisomic zygote associated with a subsequent loss of the homologue (fig 4C); and (D) mitotic misdivision at the centromere in a monosomic zygote (fig 4D). In (A) and (C), the clinical outcome might be influenced by a trisomic cell line. In some cases, the phenotype can explain the formation without any molecular investigation. For example, a normal phenotype proves postzygotic formation in der(14)t(14q;14q) or der(15)t(15q;15q),103-104 because UPD(14) and UPD(15) are associated with specific phenotypes. In other cases molecular investigations can only hint towards the formation but not delineate it definitively. In this context, one case of a cytogenetic isochromosome 8 (45,XX,−8,−8,+psu dic(8;8)(p23.3p23.3))78 and one cytogenetically similar case (45,XX,−8,−8,+psu dic(8)(p23.1p23.3))81 should be discussed. Both are heterodisomic, but in the second case haploinsufficiency of (8)(p23.1→p23.3) was found. The phenotypes are similar, but the clinical information in the second case is too sparse to correlate haploinsufficiency with specific symptoms.

Formation of UPD associated with an isochromosome. (A) Meiotic misdivision at the centromeres resulting in a gamete with an isochromosome, fertilisation by a normal gamete, and subsequent mitotic loss of the homologous chromosome. (B) Meiotic misdivision and fertilisation by a nullisomic gamete. (C) Mitotic misdivision at the centromeres of sister or non-sister chromatids in a trisomic zygote associated with a subsequent loss of the homologue. (D) Mitotic misdivision at the centromere in a monosomic zygote.
According to the molecular markers investigated, 30 out of 34 de novo cases with isochromosomes of acrocentric chromosomes are isodisomic. Despite the lack of an adequate number of investigated markers in some cases, this figure should be considered as a clue to formation by misdivision at the centromere during an early mitosis of a monosomic zygote. However, premeiotic formation was observed in one family with a maternal isodisomic dicentric isochromosome 13 in a young boy and the same, but now paternal, isodisomic dicentric isochromosome 13 in his mother.48 50 In contrast, a formation by trisomy rescue or gamete complementation, which cannot be differentiated in most cases, is more likely in heterodisomic cases, each combined with meiotic or mitotic isochromosome formation.
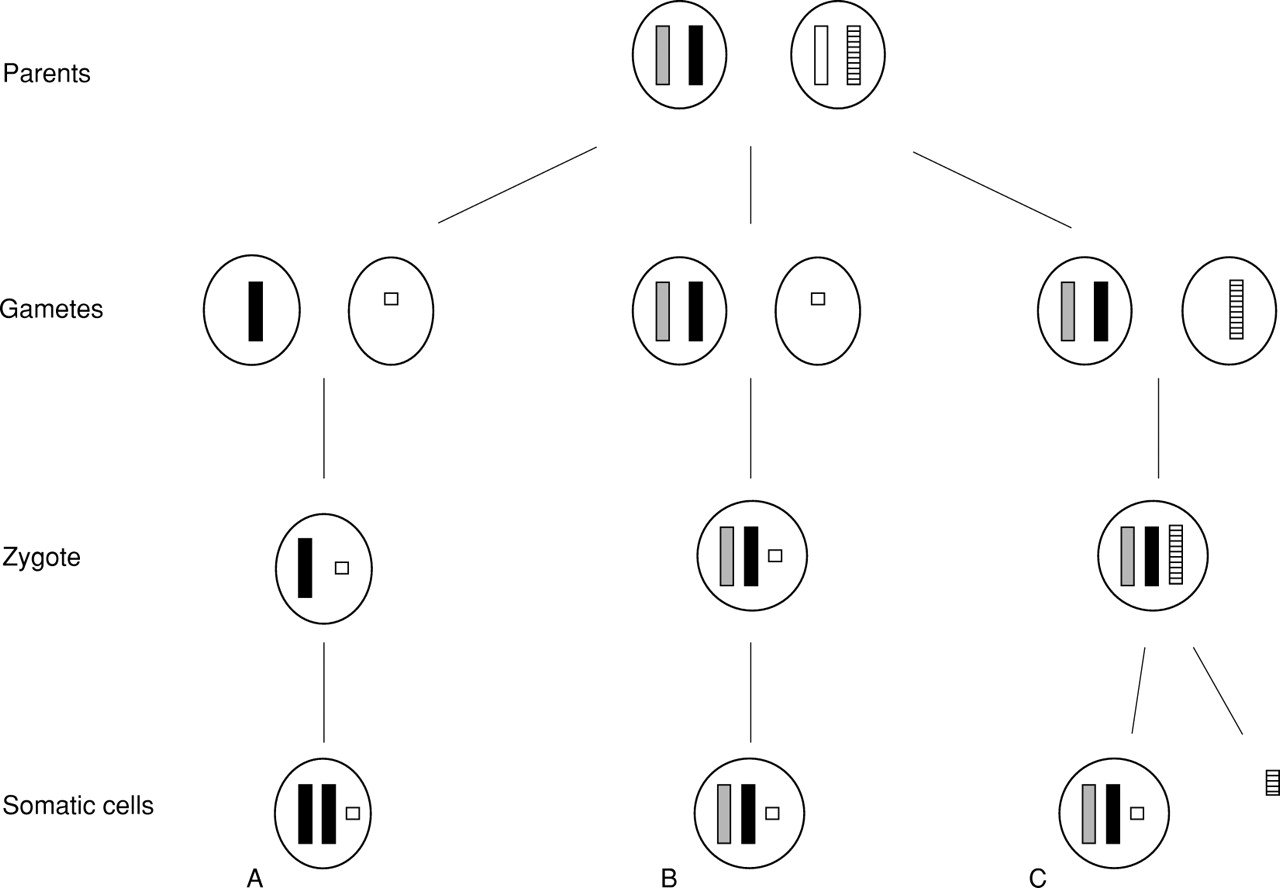
Formation of UPD associated with an additional marker chromosome will be explained mainly by two mechanisms.86 First, the marker chromosome was formed in meiosis and a gamete with the marker chromosome but no normal homologue was fertilised by a normal gamete (fig 5A). UPD arises by mitotic recombination and therefore isodisomy should always be present. Second, a disomic gamete is fertilised either by a gamete with a marker chromosome formed in meiosis (fig 5B) or by a normal gamete and subsequent mitotic formation of the marker chromosome (fig 5C). Only in the first mechanism and in the meiotic formation of a marker chromosome in the second mechanism may formation of both UPD and of the marker chromosome be related events.86 The incidence of UPD associated with an additional marker chromosome is not known. In practice, the number of cases with UPD and an associated additional marker chromosome reported so far justifies investigations for UPD, particularly if in prenatal diagnostic procedures chromosomes for which genomic imprinting is well known are found to be involved.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Formation of UPD associated with an additional marker chromosome. (A) The marker chromosome was formed in meiosis and a gamete with the marker chromosome but no normal homologue was fertilised by a normal gamete. (B) A disomic gamete is fertilised by a gamete with a marker chromosome formed in meiosis. (C) A disomic gamete is fertilised by a normal gamete and subsequent mitotic formation of the marker chromosome. In (B) and (C) not only heterodisomy as shown but also isodisomy is possible.
As shown in a case with paternal isodisomy 6, an additional r(6) chromosome in 37 out of 50 investigated cells, and transient neonatal diabetes mellitus, additional marker chromosomes can be an important clue towards mapping of genes or regions of genomic imprinting.79 However, the aetiology of clinical symptoms is difficult to understand in cases with UPD and a complex chromosomal aberration. For example, in the child with paternal uniparental isodisomy of 20p13→qter, a 45,XY,−20,−20,+ter rea(20;20)(p13p13) karyotype in lymphocytes, and a simple trisomy 20 cell line in fibroblasts, clinical anomalies might either result from the trisomic cell line, or the homozygous del(20)(p13pter) in the other cell line, or UPD of 20p13→qter.89 In addition, some case reports published recently indicate that chromosomal segregation is more complex than previously thought and therefore hidden mosaicism should always be considered.88
The risk for UPD is mostly dependent on the karyotype and its formation. For non-homologous Robertsonian translocations, Berendet al 32 estimated a risk figure for UPD of less than 1%. In contrast, homologous Robertsonian translocations such as der(14q14q) or der(15q15q) indicate a high risk for UPD. In a prospective study, four out of six homologous acrocentric rearrangements displayed UPD.32 Robinsonet al 105 estimated a theoretical risk of approximately 14% for UPD(15) in the case of a der(15q15q). In addition, there is one parent-child transmission of UPD(13) owing to an isodisomic isochromosome 13q.48 50 In cases with UPD associated with an additional marker chromosome, if one parent carries the marker chromosome, the risk for UPD may also be higher. Exact figures are not known. In contrast, the recurrence risk might be low in cases with either segmental UPD, UPD associated with a de novo true isochromosome, or UPD associated with a de novo translocation.
For phenotype-genotype correlation, three groups can be delineated; first, cases with segmental or complex UPD and no phenotypic effect, most of which were ascertained by chance. Genomic imprinting effects can be excluded for the chromosomal regions involved; second, cases with UPD(14) and (15) and the well known associated phenotypes; third, cases with an abnormal phenotype and “sensu strictu” complex UPD. Phenotype-genotype correlation in these cases is hampered by the fact that the majority of cases are unique and, in addition, a duplication and/or a deletion of genetic material may be present. Moreover, in cases with UPD and a balanced translocation apart from UPD, an additional microdeletion/duplication around the breakpoints or disruption of a gene must be taken into consideration.
Conclusion
The increasing number of cases with segmental UPD or with UPD of a part or of a whole chromosome associated with a complex chromosomal rearrangement, which have recently been described, might help in the more exact understanding of mechanisms and reasons for meiotic and/or mitotic chromosomal recombinations. In the light of these cases, it may become much easier to map genes and/or regions subject to genomic imprinting and to delineate clinical phenotypes so far undefined. Last, but not least, genetic counselling will be helped. Therefore, molecular investigations for UPD should be performed more frequently, particularly in cases with additional marker chromosomes, other complex chromosomal rearrangements, and abnormal clinical phenotypes, which cannot be explained by other causes or which are suggestive of UPD.
Note added in proof
During preparation of the manuscript another three cases with segmental UPD associated with a cytogenetically normal karyotype were published. The first described maternal segmental uniparental isodisomy of a region of around 30 cM telomeric to D4S2366 on 4p in a girl with Ellis-van Creveld syndrome.106 The second was a 3 year old boy with hyperactivity, major instability, mental retardation, facial dysmorphism, and maternal heterodisomy of a small 11 cM region of distal chromosome 17q.107 The third reported on paternal uniparental isodisomy 20q associated with a lack of the maternal specific methylation pattern within GNAS1 in a boy with PTH resistant hypocalcaemia and hypophosphataemia but without evidence for Albright hereditary osteodystrophy.108
In addition, two other case reports of complex UPD have come to my attention. First was a 45,X male patient with normal male external genitalia but clinical stigmata of Turner syndrome and azoospermia.109 UPD testing was not performed, but the laboratory results and the phenotype resembled the case reported by Weil et al.110 Second was another case with Angelman syndrome resulting from a paternal isochromosome 15.111