Article Text

Abstract
Angelman syndrome (AS) is a neurodevelopmental disorder characterised by severe mental retardation, absent speech, ataxia, sociable affect, and dysmorphic facial features. Eighty five percent of patients with AS have an identifiable genetic abnormality of chromosome 15q11-13. Mutations within the X linkedMECP2 gene have been identified in patients with Rett syndrome (RTT), a neurodevelopmental disorder which affects females almost exclusively and which shares phenotypic overlap with AS. RTT is usually associated with normal development in infancy followed by loss of acquired skills and evolution of characteristic hand wringing movements and episodes of hyperventilation.
A panel of 25 female and 22 male patients with a clinical diagnosis of AS and no molecular abnormality of 15q11-13 were screened for MECP2 mutations and these were identified in four females and one male. Following the diagnosis, it was possible to elicit a history of regression in three of these patients, who by then were showing features suggestive of Rett syndrome. In the remaining two subjects the clinical phenotype was still considered to be Angelman-like.
These findings illustrate the phenotypic overlap between the two conditions and suggest that screening forMECP2 mutations should be considered in AS patients without a demonstrable molecular or cytogenetic abnormality of 15q11-13. Since MECP2 mutations almost always occur de novo, their identification will substantially affect genetic counselling for the families concerned.
- Angelman syndrome
- Rett syndrome
- MECP2 mutations
Statistics from Altmetric.com
Angelman syndrome (AS) is a neurodevelopmental disorder characterised by severe mental retardation, inability to speak, ataxia, dysmorphic facial features, and a seizure disorder associated with a characteristic EEG appearance.1 2 Affected subjects have a specific behavioural phenotype; they are happy and sociable with a tendency to outbursts of laughter caused by minimal provocation. The subtle dysmorphic features associated with AS include deep set eyes, wide, smiling mouth, and a prominent chin. Head circumference is usually normal at birth but head growth decelerates during the early years of life and 30% of subjects become microcephalic.3 4 Most cases of AS are sporadic, although many familial cases have been reported. A variety of different genetic mechanisms involving chromosome 15q11-13, a known imprinted region, have been described in association with AS.5 Approximately 70% of patients have a de novo maternal deletion of chromosome 15q11-13, while 3% have uniparental paternal disomy for chromosome 15. In a further 5% of cases there is biparental inheritance of chromosome 15, but both chromosomes have a paternal expression pattern owing to defective imprinting of 15q11-13. Finally, around 5% of patients have loss of function mutations in the UBE3Agene, a gene encoding E6-AP, a ubiquitin protein ligase. All result in a deficiency of the E6-AP protein, which is involved in the process of ubiquitination.6 There remain a group of around 15% of AS patients, both sporadic and familial, in whom no cytogenetic or molecular abnormality involving chromosome 15q11-13 can be identified. There are few data documenting the clinical characteristics of this group of patients.7 Our own observations suggest that patients diagnosed with AS on clinical grounds alone have greater motor delay, are more likely to have some speech, and are less likely to have seizures and the characteristic Angelman syndrome EEG changes. While this group may have mutations affectingUBE3A expression which cannot be defined, there remains the possibility that AS is heterogeneous with other, as yet unidentified causes.
Rett syndrome (RTT) is a disorder which is seen almost exclusively in females.8 Classically, it is characterised by normal development for the first 12-18 months followed by developmental regression and loss of acquired skills, especially purposeful hand movement. Many girls develop characteristic “hand washing” or “hand wringing” stereotypies and abnormal breathing patterns with periods of hyperventilation and apnoea. Subjects with RTT are frequently growth retarded with acquired microcephaly and small, cold feet. Following the period of regression, skills plateau and then there is usually gradual deterioration with increasing neurological impairment over many years. The broad clinical spectrum of Rett syndrome has been described by Wan et al 9 and includes milder “formes frustes” and a severe “congenital variant” where there is no period of normal development.10 RTT is caused by mutations in the X linked gene MECP2.11 This encodes methyl CpG binding protein 2, which binds to single CpG dinucleotides throughout the genome and interacts with a histone deacetylase complex to mediate transcriptional repression. In the mouse, mecp2 is highly expressed in the brain and appears to be of importance for embryonic cellular differentiation.12
AS and RTT show phenotypic overlap, as was pointed out by Schefferet al 13 in 1990. Both are associated with severe mental retardation, acquired microcephaly, ataxia, seizures, and stereotypic hand movements. The main distinguishing feature is the clinical history as AS patients do not have an initial normal period of development or a distinct period of regression. The typical early history of regression is not present in every RTT patient and it can be difficult to elicit. We investigated the possibility that AS with no detectable abnormality of 15q11-13 may in fact result from a defect of MECP2.
Patients and methods
DNA extracted from lymphocytes of 47 patients with a clinical diagnosis of Angelman syndrome but no cytogenetic or molecular abnormality of 15q11-13 was screened for mutations within theMECP2 gene. All of these patients fulfilled the consensus diagnostic criteria for Angelman syndrome as laid down by Williams et al.14 One hundred ng DNA from each patient was amplified using the primer pairs published by Amir et al,11 which span three exons and cover the coding region of the gene. Each sample was processed through 30 cycles of amplification consisting of one minute at 94°C (denaturation), one minute at 55°C (annealing), and one minute at 72°C (extension). The final extension step was 10 minutes. SSCP/heteroduplex analysis was performed using equal volumes of PCR product and formamide loading dye. Gels were run at 350 V overnight at 4°C and silver stained according to standard protocols. They were inspected for abnormally migrating bands which were then sequenced directly using the ABI Prism Dye Terminator Cycle Sequencing kit (Perkin Elmer, Applied Biosciences Division, Warrington, UK). Results were compared to the reference sequence forMECP2 (Genebank accession No X99686.) Mutations which created or abolished restriction sites were confirmed by restriction enzyme analysis of genomic DNA using appropriate enzymes. Products of restriction digestion were fractionated by electrophoresis and the products visualised under UV transillumination after ethidium bromide staining.
Results
Mutations within MECP2 were identified in four of the 25 females and one of the 22 males screened. The clinical details of these patients are summarised in table 1 and details of the mutations identified are given in table2.
Clinical features of patients with MECP2 mutations
MECP2 mutations identified in patients 1–5
Patient 1 (fig 1) was a girl. There had been concern about her general development in the first year of life as she was a sleepy baby and sitting was delayed until 12 months of age. She had febrile seizures at 6 months and meningitis was suspected. She developed five words of speech but lost these during the second year of life. Her comprehension appeared to be better than her expressive speech. She had a small head circumference with a flat occiput and a prominent chin. She smiled frequently. She had a resting tremor and jerky voluntary movements but no hand wringing or hyperventilation. Her legs were stiff with a talipes deformity of one foot.

(A) Patient 1 at the age of 4 years and (B) at the age of 6 years.
EEG showed 2-3 Hz slow wave activity in the temporal, central, and posterior regions and some other non-specific features. A clinical diagnosis of Angelman syndrome was made. Analysis of the 15q11-13 region as described above showed no abnormalities but analysis ofMECP2 showed a 44 base pair deletion within exon 3 of the gene which was not present in either parent.
Patient 2 presented to the paediatric neurologist at the age of 3 years with severe developmental delay and ataxia. She had feeding problems as an infant and had been investigated for failure to thrive at 8 months. She sat at 7 months and her parents considered that her development had been normal up until that time. She had never been able to crawl or walk. She was noted to have a marked tremor with evidence of titubation. She was microcephalic with a flat occiput and a prominent chin and drooled frequently. She had never developed speech or had seizures but a sleep EEG was reported to be abnormal. A diagnosis of Angelman syndrome was suggested. By the age of 6 years, she had developed a scoliosis. She continued to have tremulous movements but she had no hand wringing stereotypies or hyperventilation and had some purposeful hand use. Molecular analysis of chromosome 15q11-13 was normal but analysis of MECP2 showed a C to G transition at nucleotide position 376 causing a substitution of arginine for proline at amino acid position 101, which lies within the methyl binding domain of MECP2 and would therefore be likely to have an effect on protein function.
Patient 3 was first seen at the age of 6 months because of slow developmental progress; in particular she was not reaching out for toys. At that time her head circumference was within the normal range. When seen at the age of 3 years she was noted to have frequent laughter with hand flapping and tongue thrusting behaviours and occasional fidgeting of her hands but no hand wringing. Her parents had observed heavy breathing on occasions. She was microcephalic and had a flat occiput and a prominent chin. Progress with her motor development had been slow. At 6 years she was sitting alone but not mobile. There was no history of regression. She had not had any seizures and an EEG was reported as normal. CT scan showed a large cisterna magna with a small cerebellar vermis and slightly large ventricles. A diagnosis of Angelman syndrome was suggested because of her happy affect, stereotypic hand movements, and facial features. The possibilities of Joubert syndrome and of chromosomal mosaicism were also considered. Analysis of 15q11-13 was normal, but she was found to have a 52 base pair deletion within exon 3 of MECP2.
Patient 4 was noted to have developmental delay by the health visitor at the age of 7 months. She was a floppy baby who was unable to sit without support. Records showed that early in life she was able to finger feed and transfer objects from hand to hand. Over the next three years, she had a decelerating head circumference and was noted to be brachycephalic, with facial features and a sociable disposition which were suggestive of Angelman syndrome. There was no definite history of developmental regression. On review at 6 years of age, AS was thought to be the most likely diagnosis, although she had a normal EEG and no abnormality had been detected within the 15q11-13 region. Analysis of MECP2 showed a C to G transition at position 497 leading to premature termination of the protein at amino acid position 141. This same mutation has been reported previously in girls with Rett syndrome.11
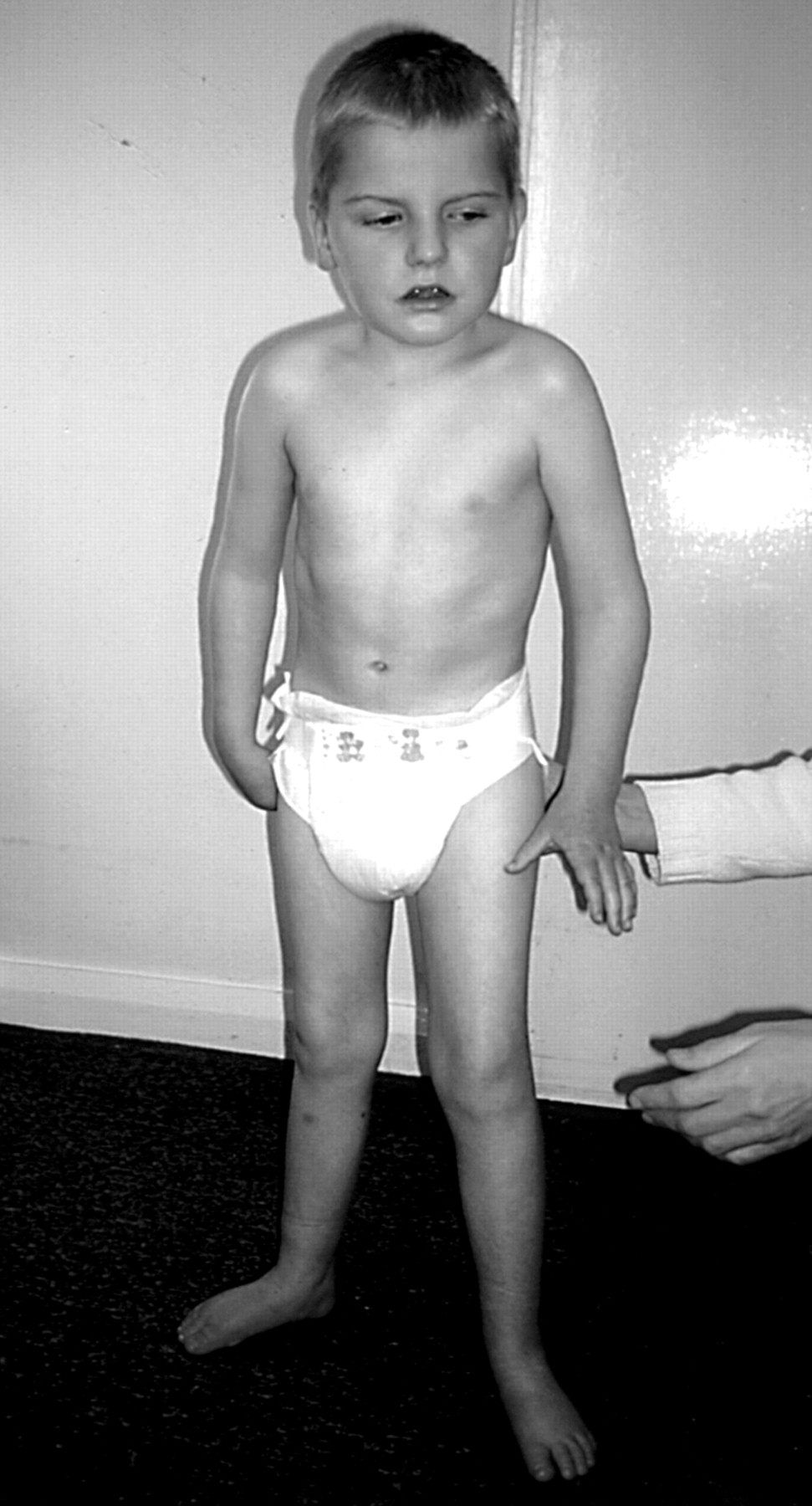
Patient 5 was a boy who had been floppy as an infant and then presented with delay in motor development during the second year of life. He walked at 15 months of age but needed a lot of support and had an unusual, wide based, stiff legged gait with marked ataxia (fig 2). In addition he had a seizure disorder and an abnormal EEG which showed an “excess of slow wave activity”. His head circumference lay between the 10th and 25th centiles but head growth had decelerated and he was brachycephalic. He drooled frequently and had some repetitive midline hand movements but no obvious hand wringing. Titubation and tremor were noted. He had reduced muscle bulk in his limbs and poor peripheral circulation with cold, blue feet. At the age of 6, he began to develop a scoliosis. The diagnosis of Angelman syndrome was made based on his facial appearance, ataxic gait, happy disposition, and seizure disorder. No abnormality of chromosome 15q11-13 was identified, but analysis of MECP2 showed that he had a 2 base pair deletion at positions 241-242 within exon 2, resulting in a frameshift with premature termination of the MeCP2 protein. The presence of a normal band on the SSCP gel suggested the possibility of somatic mosaicism. Further details of this analysis have been documented elsewhere.15 Parental analyses were normal.

{kind=link}
{kind=link}
Patient 5 at the age of 4 years. Note stiff legged posture and small, discoloured feet.
Discussion
We have identified MECP2 mutations in 4/25 girls and 1/22 boys who presented initially with a clinical diagnosis of AS. None of the subjects concerned had a demonstrable genetic abnormality of 15q11-13. The diagnosis of RTT had been considered in cases 2 and 3, as part of the differential diagnosis of AS, but RTT was not the working diagnosis in either case at the time of testing. On review of the clinical features of these five patients after identifying MECP2 mutations, four of the five had some of the classical phenotypic features of Rett syndrome which had evolved over time. These were not always typical, however, and in particular the characteristic hand washing stereotypies and hyperventilation were not prominent features. All of the five patients had the four clinical characteristics designated “consistent” (present in 100%) in the diagnostic criteria for AS laid down by Williams et al 14 in 1995. These are severe developmental delay, no or minimal use of words, movement or balance disorder, and behavioural uniqueness. In addition, all had some of the other criteria described by Williams et al 14 as being “frequent” or “associated” in AS and the clinical signs had been so convincing that it had been considered reasonable to pursue UBE3Amutation screening in each case after methylation analysis of chromosome 15 had proven normal.
It is well recognised that AS and RTT have overlapping clinical features and Scheffer et al 13 drew attention to these clinical similarities and cautioned against making the diagnosis of AS in a girl at an early age, especially in the absence of the typical EEG appearance. Laan et al 16studied the EEGs of patients with Angelman syndrome and Rett syndrome at a young age. They suggested that whereas the EEG changes in AS are pathognomonic of this condition, those seen in Rett syndrome are more non-specific and the EEG can often be normal in the first few years of life. Two of our five patients had seizures and although three had an abnormal EEG, none had the EEG features typically associated with AS. This supports the suggestion that the EEG is a good distinguishing feature. All of our patients had facial features consistent with AS and had a happy, sociable affect. Episodic laughter, often nocturnal, has been reported as a feature of Rett syndrome8 and these were frequent in the subjects we have reported. An important diagnostic feature in both AS and RTT is the presence of a movement disorder. AS is characterised by fine, tremulous movements in infancy, which evolve into coarser, ataxic voluntary movements during childhood. Hand flapping and mouthing of the hands and other objects are also common. In contrast to Rett syndrome, purposeful hand use is not well developed at any stage in AS. A typical child with Rett syndrome will initially develop purposeful hand use and will then lose this during the period of regression. From that time on, repetitive hand stereotypies such as patting, plucking clothing, tapping, hand biting, and hand wringing begin to appear, but these may be slow to evolve. In those patients where the period of regression occurs early, purposeful hand use may never develop, and in this group especially the movement disorder may be suggestive of AS in the early stages. Looking at our five patients retrospectively, prominent characteristics of the movement disorder which were distinguishable from that of AS were the presence of titubation and marked tremor.
A further observation in those of the five who were ambulant was a habit of rocking slowly and rhythmically from foot to foot. Facial features were very similar to those seen in classical AS. Three of our patients had remarkably cold, blue feet which are not a common feature of AS.
Within any condition diagnosed on clinical grounds, there remains an argument as to its exact boundaries. In view of the broad clinical variability in Rett syndrome, Hagberg10 suggested diagnostic criteria for “Rett syndrome variants” to be used in girls of 10 years or over. It is in the early years, however, that parents usually seek a diagnosis and genetic advice before planning further children. It is during this period that there is most phenotypic overlap between the features of AS and RTT. In a situation where AS is diagnosed on clinical grounds alone, parents would be counselled that recurrence risks could be as high as 50% and that no specific prenatal test would be available. If it could be shown that the child had a MECP2 mutation, the recurrence risk would be much lower in the majority of cases since most arise de novo. Clearly it is possible to argue that the patients described should not now be considered to have Angelman syndrome. The observations nevertheless serve to illustrate the potential clinical value of considering MECP2 screening in the young AS patient with no 15q11-13 abnormalities, even if the patient is a male.
The phenotypic overlap between the clinical course of AS and RTT raises the question as to whether the two conditions may be aetiologically related through a common pathogenesis. MeCP2 is involved in transcriptional silencing.17 It binds to single CpG dinucleotides throughout the genome and with the corepressor Sin3A, which binds to its transcriptional repression domain, it recruits histone deacetylases. These then act to alter chromatin configuration to prevent transcription. It has been postulated that in the presence of a MECP2 mutation, there may be overexpression of downstream genes, although a study by Xianget al 18 did not show an increase in levels of certain neurotransmitters within the brains of subjects with MeCP2 mutations, which would have supported this hypothesis. Overexpression of genes within the 15q11-13 region does give rise to a specific phenotype. Several subjects with a maternal duplication of this region have had autistic features, ataxia, epilepsy, learning difficulties, and subtle dysmorphic facial features.19 20One hypothesis, therefore, is that UBE3A is one of the target genes downstream of MECP2for which alteration of expression is particularly critical. Further investigation of this hypothesis will involve the study of those areas of the brain where UBE3A is known to be expressed.
Acknowledgments
GB is a Clinician Scientist Fellow funded by the Wellcome Trust (reference 51390/Z). PW is funded by a grant from the Birth Defects Foundation.